Воспаление — словарь патологии — MyPathologyReport.ru
Что значит воспаление?
Воспаление — это естественная защита организма от травм или болезней. Организм также использует этот процесс для восстановления тканей после травмы. Особые клетки, участвующие в воспалении, называются воспалительные клетки и они являются частью иммунной системы организма.
Стадии воспаления
Острый воспалениеВоспаление делится на две стадии. Первый этап называется острое воспаление. Эта стадия начинается вскоре после травмы и обычно длится всего пару дней. Однако эта стадия может продолжаться и дольше, если причина травмы или болезни не исчезнет. Например, острое воспаление, вызванное инфекцией, может продолжаться до тех пор, пока вызывающий инфекцию микроорганизм все еще находится в организме. В некоторых частях тела патологи используют слово «активный» для описания этой стадии.
Типы иммунных клеток, которые принимают участие в этой стадии, включают нейтрофилы и эозинофилы.
Второй этап называется хроническое воспаление. Не все травмы или заболевания вызывают хроническое воспаление. Когда эта стадия действительно возникает, она обычно начинается сразу после окончания острой воспалительной стадии. Эта стадия может длиться несколько дней или недель в зависимости от травмы или заболевания.
Типы иммунных клеток, которые принимают участие в хроническом воспалении, включают: плазменные клетки, лимфоциты, и гистиоциты.
Изменения, вызванные воспалением
Хотя воспаление помогает организму бороться с болезнью и восстанавливать после травмы, воспалительная реакция также может вызвать повреждение близлежащих клеток. Патологи часто описывают клетки, поврежденные воспалительным процессом, как показывающие реактивное изменение.
Заболевания, вызванные воспалением
Некоторые заболевания вызваны воспалением, которое продолжается длительное время. Воспалительные клетки повреждают нормальные клетки и ткани, и это мешает правильному функционированию органа.
Примеры включают в себя:
- Воспалительные заболевания кишечника (Крона и язвенный колит).
- Целиакия.
- Ревматоидный артрит.
Хроническая обструктивная болезнь легких (ХОБЛ)
Основные факты
- Хроническая обструктивная болезнь легких (ХОБЛ) – третья причина смерти по всем мире, от которой в 2019 г. умерло 3,23 млн человек
- Более 80% этих случаев смерти приходятся на долю стран с низким и средним уровнем дохода (СНСД).
- ХОБЛ вызывает стойкие и прогрессирующие респираторные симптомы, к которым относятся одышка, кашель и/или выделение мокроты.
- Причиной развития ХОБЛ является хроническое воздействие вредных газов и мелкодисперсных частиц в сочетании с индивидуальными факторами, в том числе обстоятельствами, влияющими на развитие легких в детстве, а также генетическими факторами.
- Важными факторами риска развития ХОБЛ являются воздействие табачного дыма, загрязнение воздуха в помещениях, а также вдыхание пыли, дыма и химических веществ на рабочем месте.

- Для замедления прогрессирования симптомов и уменьшения частоты обострений необходимы ранняя диагностика и лечение, включая помощь в отказе от курения.
Что такое ХОБЛ?
ХОБЛ – распространенное, предотвратимое и поддающееся лечению хроническое заболевание легких, от которого страдают как мужчины, так и женщины во всем мире.
Поражение мелких дыхательных путей в легких приводит к нарушению входящего и выходящего воздушного потока. Сужение просвета дыхательных путей является следствием ряда процессов. Могут иметь место частичное разрушение тканей легкого, облитерация дыхательных путей мокротой, воспалительный процесс и отек слизистой оболочки дыхательных путей.
Иногда для обозначения ХОБЛ используют термины «эмфизема» или «хронический бронхит». Как правило, понятие «эмфизема» означает разрушение мелких воздушных полостей легкого, которыми заканчиваются дыхательные пути. Хронический
бронхит – это хронический кашель с выделением мокроты, вызванный воспалением дыхательных путей.
У ХОБЛ и астмы есть ряд общих симптомов (кашель, свистящее дыхание и одышка), и оба заболевания могут протекать параллельно.
Распространенные симптомы ХОБЛ начинают проявляться в среднем возрасте и включают в себя:
- одышку или затрудненное дыхание;
- хронический кашель, часто с выделением мокроты; и/или
- утомляемость.
По мере прогрессирования ХОБЛ больному становится все труднее выполнять нормальные повседневные действия, часто вследствие одышки. Связанные с этим снижение производительности труда и бытовые ограничения, а также расходы на лечение приводят к значительному финансовому бремени.
Во время обострений симптомы ХОБЛ становятся намного более тяжелыми, в связи с чем больным может требоваться дополнительное лечение на дому или госпитализация для оказания экстренной медицинской помощи. Тяжелые обострения могут угрожать жизни больного.
Часто у людей, страдающих ХОБЛ, отмечаются сопутствующие заболевания, такие как сердечно-сосудистые заболевания, остеопороз, нарушения опорно-двигательной системы, рак легкого, депрессия и тревожные расстройства.
Для ХОБЛ характерно постепенное развитие, часто в результате воздействия комбинации факторов риска:
- вдыхание табачного дыма при активном и пассивном курении;
- профессиональное ингаляционное воздействие пыли, дыма или химических веществ;
- загрязнение воздуха в помещениях: в странах с низким и средним уровнем дохода при приготовлении пищи или для отопления часто применяются топливо на основе биомассы (древесина, навоз, пожнивные остатки) или уголь, с чем связан высокий уровень воздействия дыма в жилых помещениях;
- особенности первых лет жизни, например нарушения внутриутробного развития, недоношенность, а также частые или тяжелые респираторные инфекции в детском возрасте, препятствующие полноценному росту и развитию легких;
- астма в детском возрасте; и/или
- редкое генетическое нарушение – дефицит альфа-1-антитрипсина – может привести к развитию ХОБЛ в раннем возрасте.

Снижение бремени ХОБЛ
ХОБЛ не поддается излечению, однако ранняя диагностика и лечение очень важны и могут помочь замедлить прогрессирование симптомов и снизить риск обострений.
Болезнь можно заподозрить при наличии характерных симптомов, и для подтверждения диагноза проводятся функциональные дыхательные пробы, называемые спирометрией, которые позволяют измерить работу легких. В странах с низким и средним уровнем дохода часто возможности для проведения спирометрии отсутствуют, в результате чего заболевание может остаться недиагностированным.
Больные ХОБЛ могут принять ряд мер для улучшение своего общего состояния и сдерживания прогрессирования заболевания:
- прекращение курения – лицам с ХОБЛ следует обеспечить помощь в отказе от курения;
- регулярная физическая активность; и
- вакцинация против пневмонии, гриппа и коронавирусной инфекции.
Для облегчения симптомов и снижения интенсивности обострений могут назначаться ингаляционные препараты. Существует множество различных ингаляционных препаратов с разным механизмом действия, некоторые из которых могут быть комбинированными.
Существует множество различных ингаляционных препаратов с разным механизмом действия, некоторые из которых могут быть комбинированными.
Некоторые ингаляционные препараты способствуют расширению дыхательных путей; они могут назначаться как на регулярной основе в целях предупреждения или смягчения симптомов, так и для облегчения симптомов во время сильных обострений. Иногда для снижения воспаления тканей легких в комбинации с этими препаратами назначаются ингаляционные кортикостероиды.
Использование ингаляционных препаратов требует правильной техники, и в некоторых случаях применяются так называемые спейсерные устройства, позволяющие более эффективно доставлять препарат в дыхательные пути. Во многих странах с низким и средним уровнем дохода доступ к ингаляционным препаратам ограничен; так, в 2019 г. ингаляционный сальбутамол имелся в наличии в лечебных учреждениях первичного звена только в половине стран с низким уровнем дохода [2].
Нередко обострения бывают спровоцированы респираторной инфекцией; в этих случаях в дополнение к ингаляционным препаратам или небулайзерам по мере необходимости могут назначаться антибиотики и/или пероральные стероидные препараты.
Людям, живущим с ХОБЛ, следует предоставлять информацию об их заболевании, необходимом лечении и правилам оказания самопомощи, что поможет им вести максимально активную и здоровую жизнь.
Стратегия ВОЗ по профилактике и контролю ХОБЛПроблема ХОБЛ отражена в Глобальном плане действий ВОЗ по профилактике неинфекционных заболеваний (НИЗ) и борьбе с ними и в Повестке дня Организации Объединенных Наций в области устойчивого развития на период до 2030 г.
ВОЗ предпринимает ряд мер для расширения диагностики и лечения ХОБЛ.
Для помощи в борьбе с НИЗ на уровне первичной медико-санитарной помощи в условиях ограниченных ресурсов ВОЗ разработала комплекс основных мер борьбы с неинфекционными заболеваниями. Этот комплекс включает в себя протоколы для оценки, диагностики и ведения
хронических респираторных заболеваний (астмы и хронической обструктивной болезни легких), а также модули, касающиеся консультирования по вопросам здорового образа жизни, включая отказ от курения и самопомощь.
«Реабилитация 2030» – это новый стратегический подход, призванный способствовать приоритетному развитию и укреплению реабилитационных услуг в рамках систем здравоохранения. В настоящее время в рамках этой инициативы ВОЗ разрабатывает комплекс мероприятий по реабилитации, который кроме прочего включает в себя легочную реабилитацию при ХОБЛ.
Снижение воздействия табачного дыма крайне важно как для первичной профилактики ХОБЛ, так и в контексте ведения больных. Прогрессу в этой области способствует Рамочная конвенция по борьбе против табака, а также такие инициативы ВОЗ, как MPOWER и mTobacco Cessation.
К числу других действий в сфере профилактики относится разработка Набора инструментов для обеспечения чистой энергии в быту (CHEST), который направлен на продвижение чистых и безопасных бытовых энергетических технологий и содействие разработке политики,
способствующей внедрению чистых источников энергии в быту на местном, программном и национальном уровнях.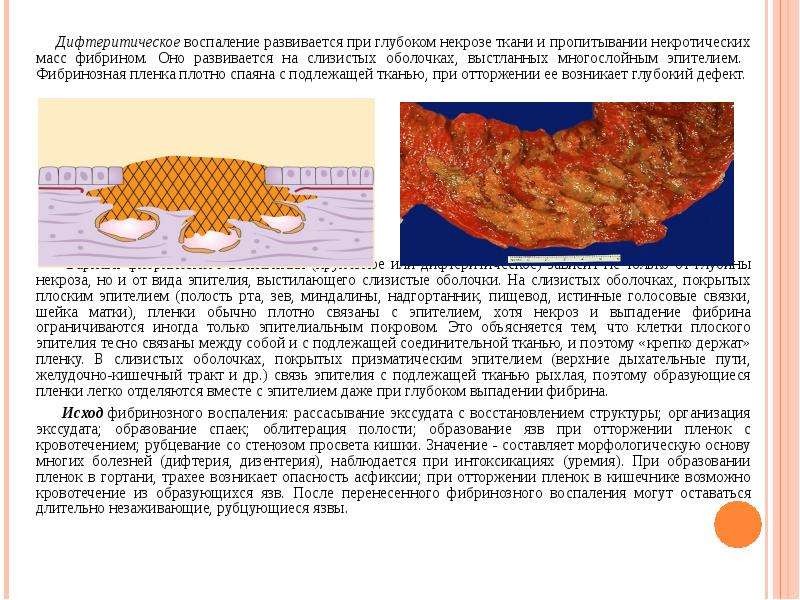
Глобальный альянс по борьбе с хроническими респираторными болезнями (GARD) содействует работе ВОЗ по профилактике и контролю хронических респираторных заболеваний. GARD представляет собой широкий добровольный альянс национальных и международных организаций и учреждений, работающих над достижением общей цели по уменьшению глобального бремени хронических респираторных болезней.
Библиография
1. WHO Global Health Estimates
2. Assessing national capacity for the prevention and control of noncommunicable diseases: report of the 2019 global survey. Geneva: World Health Organization; 2020. Licence: CC BY-NC-SA 3.0 IGO
Лечение воспаления шейки матки (цервицит) на любой стадии в Приморском районе СПб
Воспаление шейки матки (цервицит) – воспаление такни внутренней стенки матки (эндометрия). Как правило, сопровождается инфекциями, которые переносятся половым путём, например, хламидиоз, трихомониаз, генитальный герпес, гонорея.
ЛЕЧЕНИЕ ВОСПАЛЕНИЯ ШЕЙКИ МАТКИ ДОСТУПНО В ФИЛИАЛАХ:
Лечение воспаления шейки матки в Приморском районе
Адрес: г. Санкт-Петербург, Приморский район, ул. Репищева, 13
Лечение воспаления шейки матки в Петроградском районе
Адрес: г. Санкт-Петербург, Петроградский район, ул. Ленина, 5
Лечение воспаления шейки матки во Всеволожске
Адрес: г. Всеволожск, Октябрьский пр-т, 96 А
Симптомы воспаления шейки матки
Симптомами воспаления шейки матки, вызванного, например, возбудителями хламидиями, являются:
- прозрачные или желтоватые выделения из влагалища,
- зуд,
- покраснения,
- может возникать боль, во время мочеиспускания.
Кроме половых инфекций причиной воспаления могут стать различные патогены, такие как кишечная палочка, стрептококки, стафилококки, гонококки.![]()
Симптоматика заболевания часто бывает смазана и из-за этого женщина может длительное время не видеть проблему и не обращаться к врачу.
В некоторых случаях причиной воспаления шейки матки могут быть различные травмы, в том числе опущения влагалища, разрыв шейки матки во время родов. Воспаления могут возникать из-за инфекций, проникших вследствие абортов и диагностических выскабливаний. Также считается, что воспалительные процессы в шейке матки могут возникать из-за аллергии на средства интимной гигиены, латексные презервативы.
Так же причиной воспалительных процессов может стать ослабление со стороны иммунной системы.
Диагностика воспаления шейки матки
Воспаление влагалищной части шейки матки — экзоцервикса, называется экзоцервицит, и при обследовании выявляется как покраснение слизистой оболочки, которая покрывает эту часть шейки матки. Воспаление канала шейки матки называется эндоцервицитом.
Постановка диагноза воспаление шейки матки, обычно сопровождается определением степени сложности заболевания:
- Острое воспаление шейки матки — инфекция в организме свежая и активно развивающаяся
- Хроническое воспаление шейки матки – возникает, если инфекция, вызвавшая воспаление шейки, не излечена и на протяжении длительного времени разрушает ткани.
- Гнойное воспаление шейки матки – обозначает, что воспаление сопровождается нагноением в канале шейки матки, наличие гнойного цервицита среди будущих мам увеличивает риск нарушения нормального течения всей беременности и последующих родов
- Вирусное воспаление шейки матки – причина воспаления — вирус, передающийся половым путем
- Бактериальное воспаление шейки матки – причина воспаления — бактериальная инфекция (гонорея, бактериальный вагиноз)
- Кандидозное воспаление шейки матки – причина воспаления — грибковая инфекция;
- Неспецифическое воспаление шейки матки – воспаление шейки возникло в результате изменения микрофлоры.
- Кистозное воспаление шейки матки — причина заболевания – сочетание вирусного и бактериального поражения, что приводит к разрастанию цилиндрического эпителия (клетки цилиндрической формы, входящие в состав слизистой оболочки) вдоль поверхности матки, что приводит к образованиям кист.
- Атрофическое воспаление шейки матки – воспаление сопровождается истончением тканей шейки матки (при хронической форме цервицита, эрозии шейки матки, циститах, воспалении придатков)
- Очаговое воспаление шейки матки – воспаление проявляется на отдельных участках слизистой шейки матки.

Воспаления шейки матки при беременности
Наличие воспаления шеки матки при беременноси повышает риск осложнения протекания родов. Само воспаление, как и вызвавшая его болезнь, могут стать причиной преждевременных родов, инфицировании плода и развития у него различных форм заболеваний.
На ранних сроках беременности воспалительные процессы могут привести к замиранию плода и выкидыша. На поздних стадиях – к задержке внутриутробного развития и инфицирования ребёнка.
Возможность забеременнеть при наличии воспаленией шейки матки так же не высока, так заболевание является комплексным и затрагивает всю половую систему женщины.
Лечение воспаления шейки матки
Особое внимание необходимо обратить на то, что отсутствие лечения воспаления шейки матки может приводить к появлению эрозий и разнесению инфекции по верхним отделами половых органов.
При подборе лечения воспаления матки учитывается стадия воспаления, поставленная при осмотре врачом.
Для лечения воспалительных процессов применяются противовирусные, антибактериальные и иные средства, принимая во внимание причины заболевания, природу возбудителя и его чувствительности к выбранному препарату. Так же возможно использование местных препаратов: кремов и свечей.
При хронических стадиях заболевания могут потребоваться хирургические вмешательства (такие как, лазеротерапии, криотерапии, диатермокоагуляции).
Очень важно отметить, что вместе с лечением воспаления шейки матки необходимо лечить и сопутствующие заболевания. А для эффективности лечения необходимо параллельно обследование и лечение полового партнёра.
Если возникает подозрение на воспалительные процессы в шейке матки, а так же при наличии характерных симптомов (очень болезненные пмс), следует обратиться к гинекологу. Кроме того, может понадобиться консультация уролога.
Не ждите, что заболевание пройдёт само! Не занимайтесь самолечением!!
В нашей клинике вы можете пройти полное обследование и необходимый курс лечения на современном уровне, получить рекомендации и советы высококвалифицированных специалистов.
Прием ведут врачи:
Выберите филиал“Династия” на Новочеркасском пр-те, Красногвардейский район“Династия” на Ленина, Петроградский район“Династия” на Репищева, Приморский район“Династия” во ВсеволожскеВыездная служба
Стоимость лечения воспаления шейки матки:
| Наименование услуг | Цена в рублях | |
| Санкт-Петербург | Всеволожск | |
| Первичный прием акушера-гинеколога 1 ступени | 1850 | 1500 |
| Повторный прием акушера-гинеколога 1 ступени | 1650 | 1300 |
| Первичный прием акушера-гинеколога 2 ступени | 2100 | — |
| Повторный прием акушера-гинеколога 2 ступени | 1900 | — |
| МАНИПУЛЯЦИИ | ||
| Введение акушерского пессария | 1500 | 1500 |
| Введение внутриматочной спирали (ВМС) | 2500 | 2500 |
| Введение внутриматочной спирали «Мирена» | 4000 | 4000 |
| Введение имплантируемого контрацепива «ИМПЛАНОН» (без стоимости контрацептива) | 2500 | 2100 |
| Видеокольпоскопия | 1700 | 1700 |
| Забор мазков (гинекологический) | 300 | 250 |
| Инструментальное удаление внутриматочной спирали (ВМС) | 2500 | 2100 |
| Интравагинальное введение свечей (без стоимости медикаментов), 1 процедура | 500 | 500 |
| Лечебная обработка влагалища | 700 | 700 |
| Медикаментозное прерывание беременности | 8000 | — |
| Местная обработка наружных половых органов | 700 | 700 |
| Удаление акушерского пессария | 1000 | 1000 |
| Удаление внутриматочной спирали (ВМС) | 1500 | 1500 |
| Удаление внутриматочной спирали «Мирена» | 3000 | 3000 |
| Удаление имплантируемого контрацепива «ИМПЛАНОН» | 2500 | 2300 |
| Удаление инородного тела из влагалища | 1800 | 1800 |
| Штрих-биопсия эндометрия (пайпель-диагностика) | 1500 | 1200 |
| PRP-терапия | — | 3500 |
| УЛЬТРАЗВУКОВАЯ И ФУНКЦИОНАЛЬНАЯ ДИАГНОСТИКА | ||
| УЗИ органов малого таза (одним датчиком) | 1600 | 1300 |
| УЗИ органов малого таза (двумя датчиками) | 1900 | 1500 |
| КТГ (кардиотокография плода) | 1800 | — |
| КТГ (многоплодная беременность) | 2300 | — |
| ЛАЗЕРНАЯ ХИРУРГИЯ | ||
| Биопсия шейки матки + гистология | 5000 | 4000 |
| Единичная киста шейки матки | 1500 | 1500 |
| Единичные папилломы и кондиломы стенок влагалища | 3000 | 3000 |
| Распространенный кондиломатоз, папилломатоз | от 6000 | от 6000 |
Удаление единичных кондилом, папиллом вульвы, шейки матки (за 1 ед. ) ) |
600 | 600 |
| Эктопия (эрозия) шейки матки (менее 2 см) | 6000 | 4000 |
| Эктопия (эрозия) шейки матки (более 2 см) | 9000 | 9000 |
| Эндометриоз шейки матки (единичный очаг) | 1000 | 1000 |
| РАДИОХИРУРГИЯ | ||
| Биопсия шейки матки радионожом | 2500 | 2000 |
| Лечение лейкоплакии и крауроза вульвы с помощью радиохирургического ножа | 5300 | 5100 |
| Лечение патологии шейки матки с помощью радиохирургического ножа до 1 см | 5000 | 3700 |
| Лечение патологии шейки матки с помощью радиохирургического ножа до 2 см | 6500 | 6000 |
| Удаление полипов шейки матки с помощью радиохирургического ножа | 4000 | 2500 |
| Фульгурация кист, эндометриоидных очагов с помощью радиохирургического ножа | 3500 | 2200 |
Электрокоагуляция кондилом на коже в области промежности радионожом (за 1 ед. ) ) |
800 | 800 |
| Электрокоагуляция кондилом, папиллом вульвы радионожом | 1400 | 1400 |
| Электрокоагуляция кондилом, папиллом влагалища радионожом | от 3200 | от 3200 |
| ОПЕРАЦИИ | ||
| Интимная контурная пластика | — | 26000 |
| Хирургическая дефлорация | 8000 | 8000 |
ЗАПИСЬ НА ЛЕЧЕНИЕ ВОСПАЛЕНИЯ ШЕЙКИ МАТКИ
Ваша заявка отправлена
Менеджер свяжется с вами для уточнения деталей
Мы ценим ваше обращение в наш медицинский центр «Династия»
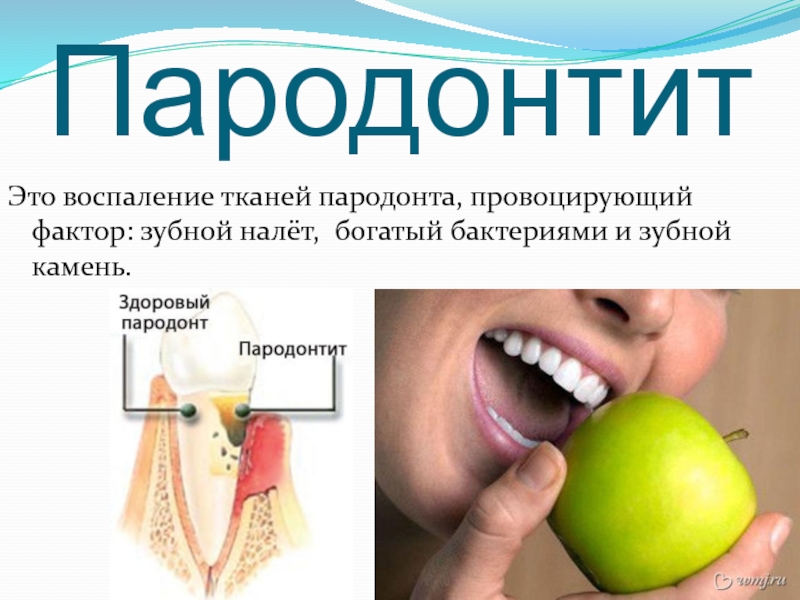
Воспаление десен | Без боли
Природные условия Санкт-Петербурга нелегкое испытание для человеческого иммунитета. Перепады температуры, высокая влажность, недостаток ультрафиолета, вода «бедна» микроэлементами и минералами необходимыми организму все это способствует подрыву иммунитета.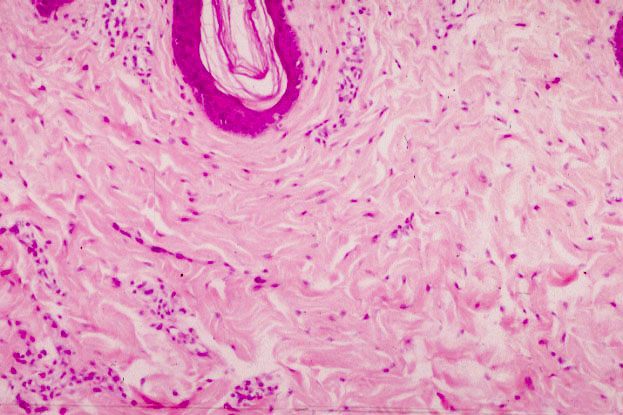 Одно из самых коварных заболеваний нашего региона воспаление десен — пародонтит.
Одно из самых коварных заболеваний нашего региона воспаление десен — пародонтит.
Как правило, люди оставляют без внимания первые признаки воспаления десен:
Легкая припухлость
Покраснение всей десны или отдельного участка
Небольшая болезненность и дискомфорт при чистке зубов
Однако, такие симптомы – тревожный признак начинающегося гингивита – воспаления околозубных тканей. Если не провести лечение вовремя, то гингивит может перерасти в пародонтит.При пародонтите появляется кровоточивость десен, появляются так называемые над- и поддесневые карманы – места, где ткань десны отходит от зуба. В десневых карманах остаются частички пищи, служащие великолепной средой для размножения болезнетворных микроорганизмов, ускоряющих воспалительные процессы в околозубных тканях. При запущенном пародонтите инфекция может поразить костную ткань, что приведет к ее размягчению, и в этом случае пациент потеряет вполне здоровый зуб.
Неизбежным спутником гингивита и пародонтита является галитоз – неприятный запах изо рта, вызываемый продуктам жизнедеятельности микробов.
-
- Чаще всего воспаление десен является результатом неправильной или недостаточной гигиены рта, однако, когда иммунитет неизбежно ослабевает, оно вполне может возникнуть у людей, ухаживающих за своими зубами тщательно.
-
- Способствовать воспалению десен могут вирусные инфекции дыхательных путей, обострение хронического гайморита, синусита и другие лор-заболевания, а так же курение, употребление алкоголя, привычка дышать ртом.
-
- Способствовать воспалению десен могут и гормональные сдвиги в организме, в том числе циклические, например, менструация у женщин или климакс.
Запишитесь на консультацию к пародонтологу
В среднем, запущенный гингивит перерастает в пародонтит в срок от полугода до трех лет, в зависимости от состояния организма и индивидуальных особенностей человека.
Поэтому необходимо посещать стоматолога-пародонтолога не реже одного раза в полгода, желательно в опасный период межсезонья. Так же следует незамедлительно обратиться к врачу при обнаружении первых признаков воспаления десен, описанных выше. Врач поможет установить причины инфекции и устранит, по возможности, ее источник.
Так же следует незамедлительно обратиться к врачу при обнаружении первых признаков воспаления десен, описанных выше. Врач поможет установить причины инфекции и устранит, по возможности, ее источник.
Полезные статьи
Домашний уход за зубами
Защита межзубных промежутков
Как вернуть 13% за лечение
Профессиональная гигиеническая чистка
Методы лечения людей с некротическим панкреатитом (разрушение поджелудочной железы вследствие eё воспаления)
Вопрос обзора
Как следует лечить людей с некротическим панкреатитом?
Актуальность
Поджелудочная железа — это орган в брюшной полости (животе), который секретирует несколько пищеварительных ферментов (вещества, которые обеспечивают и ускоряют химические реакции в организме) в протоковую систему поджелудочной железы, которые выделяются в тонкий кишечник. Она также содержит островки Лангерганса, которые секретируют несколько гормонов, включая инсулин (помогает регулировать сахар в крови). Острый панкреатит — внезапное воспаление поджелудочной железы, которое приводит к ее разрушению (некроз поджелудочной железы.) Некроз поджелудочной железы может быть инфицированным или неинфицированным (стерильным). Некроз поджелудочной железы может приводить к недостаточности других органов, таких как легкие и почки, и является жизнеугрожающим заболеванием. Основные виды лечения некроза поджелудочной железы включают в себя удаление мертвой ткани (удаление некротических тканей или некрэктомия), перитонеальный лаваж (вымывание мертвых тканей из брюшной полости), дренаж (установка трубки или «дренажа» для удаления скапливающейся вокруг поджелудочной железы жидкости), или первичный дренаж с последующей некрэктомией, при необходимости (так называемый, минимально инвазивный поэтапно возрастающий [«step-up»] подход). Минимально инвазивный поэтапно возрастающий подход может быть выполнен разными способами. Например, при минимально инвазивном поэтапно возрастающем подходе с видео-ассистированием, некрэктомия производится после периода дренирования с помощью операции через минимальный доступ, некрэктомия производится с помощью эндоскопа (инструмент, используемый, чтобы посмотреть внутрь брюшной полости).
Острый панкреатит — внезапное воспаление поджелудочной железы, которое приводит к ее разрушению (некроз поджелудочной железы.) Некроз поджелудочной железы может быть инфицированным или неинфицированным (стерильным). Некроз поджелудочной железы может приводить к недостаточности других органов, таких как легкие и почки, и является жизнеугрожающим заболеванием. Основные виды лечения некроза поджелудочной железы включают в себя удаление мертвой ткани (удаление некротических тканей или некрэктомия), перитонеальный лаваж (вымывание мертвых тканей из брюшной полости), дренаж (установка трубки или «дренажа» для удаления скапливающейся вокруг поджелудочной железы жидкости), или первичный дренаж с последующей некрэктомией, при необходимости (так называемый, минимально инвазивный поэтапно возрастающий [«step-up»] подход). Минимально инвазивный поэтапно возрастающий подход может быть выполнен разными способами. Например, при минимально инвазивном поэтапно возрастающем подходе с видео-ассистированием, некрэктомия производится после периода дренирования с помощью операции через минимальный доступ, некрэктомия производится с помощью эндоскопа (инструмент, используемый, чтобы посмотреть внутрь брюшной полости).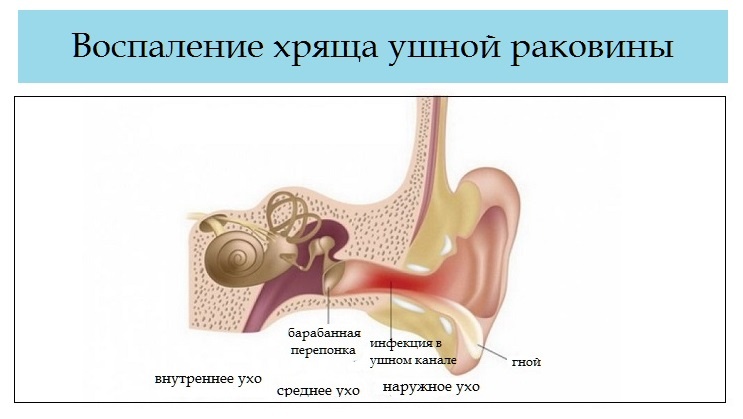
Лучший способ лечения людей с некротическим панкреатитом не ясен. Мы стремились решить эту проблему путем поиска существующих исследований на эту тему. Мы включили все рандомизированные контролируемые испытания (клинические исследования, где люди случайным образом распределены в одну из двух или более групп лечения), результаты которых были опубликованы до 7 апреля 2015 года.
Характеристика исследований
Восемь испытаний, включающие 311 участников, соответствовали критериям включения в обзор, из которых 306 участников были включены в различные сравнения. Лечение, сравнивавшееся в пяти испытаниях, включало некрэктомию, перитонеальный лаваж и поэтапно возрастающий подход. Три других испытания сравнивали вариации в сроках некрэктомии и методах поэтапно возрастающего подхода. Участники испытаний имели инфицированный или стерильный некроз поджелудочной железы в результате различных причин.
Основные результаты
В целом, уровень краткосрочной смертности (смертность в течение короткого времени) был 30% и уровень серьезных неблагоприятных событий (побочных эффектов или осложнений) был 139 на 100 участников. Различия в краткосрочной смертности или в проценте людей с серьезными неблагоприятными событиями были неточными во всех сравнениях. Число серьезных неблагоприятных событий и неблагоприятных событий было меньше при минимально инвазивном поэтапно возрастающем подходе по сравнению с открытой некрэктомией. Осложнения в результате заболевания и лечения включали сердечную недостаточность (сердце не перекачивает достаточно крови по телу при соответствующем давлении), легочная недостаточность (легкие не удаляют продукты жизнедеятельности из крови) и заражение крови (микроорганизмы и их отравляющие вещества в крови). Процент людей с недостаточностью органов и средняя стоимость были ниже при минимально инвазивном поэтапно возрастающем подходе по сравнению с открытой некрэктомией. Число неблагоприятных событий было больше при минимально инвазивном поэтапно возрастающем подходе с видео ассистированием по сравнению с минимально инвазивным поэтапно возрастающим подходом с эндоскопическим ассистированием, но общее число выполненных процедур было меньше при минимально инвазивном поэтапно возрастающем подходе с видео ассистированием по сравнению с эндоскопическим минимально инвазивным возрастающим подходом.
Различия в краткосрочной смертности или в проценте людей с серьезными неблагоприятными событиями были неточными во всех сравнениях. Число серьезных неблагоприятных событий и неблагоприятных событий было меньше при минимально инвазивном поэтапно возрастающем подходе по сравнению с открытой некрэктомией. Осложнения в результате заболевания и лечения включали сердечную недостаточность (сердце не перекачивает достаточно крови по телу при соответствующем давлении), легочная недостаточность (легкие не удаляют продукты жизнедеятельности из крови) и заражение крови (микроорганизмы и их отравляющие вещества в крови). Процент людей с недостаточностью органов и средняя стоимость были ниже при минимально инвазивном поэтапно возрастающем подходе по сравнению с открытой некрэктомией. Число неблагоприятных событий было больше при минимально инвазивном поэтапно возрастающем подходе с видео ассистированием по сравнению с минимально инвазивным поэтапно возрастающим подходом с эндоскопическим ассистированием, но общее число выполненных процедур было меньше при минимально инвазивном поэтапно возрастающем подходе с видео ассистированием по сравнению с эндоскопическим минимально инвазивным возрастающим подходом. Различия в каких-либо других сравнениях по числу серьезных неблагоприятных событий, процент людей с недостаточностью органов, число неблагоприятных событий, длительность пребывания в больнице и пребывание в отделении интенсивной терапии были либо неточными, либо непоследовательными (несогласованными). Ни одно из испытаний не сообщало о долгосрочной смертности, инфицированном некрозе поджелудочной железы (в испытаниях, которые включали участников со стерильным некрозом), качестве жизни, связанном со здоровьем (которое измеряет физическое, умственное, эмоциональное и социальное функционирование), о проценте людей с неблагоприятными событиями, о необходимости дополнительных инвазивных вмешательств, о времени возвращения к нормальной активности и времени возвращения к работе.
Различия в каких-либо других сравнениях по числу серьезных неблагоприятных событий, процент людей с недостаточностью органов, число неблагоприятных событий, длительность пребывания в больнице и пребывание в отделении интенсивной терапии были либо неточными, либо непоследовательными (несогласованными). Ни одно из испытаний не сообщало о долгосрочной смертности, инфицированном некрозе поджелудочной железы (в испытаниях, которые включали участников со стерильным некрозом), качестве жизни, связанном со здоровьем (которое измеряет физическое, умственное, эмоциональное и социальное функционирование), о проценте людей с неблагоприятными событиями, о необходимости дополнительных инвазивных вмешательств, о времени возвращения к нормальной активности и времени возвращения к работе.
Качество доказательств
Общее качество доказательств было низким или очень низким для всех измерений, потому что испытания были с высоким риском смещений (например, предубеждение людей, которые проводили испытание и участники испытаний, которые предпочитали один вид лечения другому), и испытания были малыми. В результате, необходимы дальнейшие исследования по этой теме.
В результате, необходимы дальнейшие исследования по этой теме.
Воспалительные реакции и заболевания органов, связанные с воспалением
Резюме
Воспаление — это биологический ответ иммунной системы, который может быть вызван множеством факторов, включая патогены, поврежденные клетки и токсичные соединения. Эти факторы могут вызывать острые и / или хронические воспалительные реакции в сердце, поджелудочной железе, печени, почках, легких, головном мозге, кишечном тракте и репродуктивной системе, потенциально приводя к повреждению тканей или заболеванию. И инфекционные, и неинфекционные агенты, а также повреждение клеток активируют воспалительные клетки и запускают воспалительные сигнальные пути, чаще всего пути NF-κB, MAPK и JAK-STAT.Здесь мы рассматриваем воспалительные реакции внутри органов, уделяя особое внимание этиологии воспаления, механизмам воспалительного ответа, разрешению воспаления и органоспецифическим воспалительным ответам.
Ключевые слова: воспаление, воспалительные сигнальные пути, хемокины, цитокины, заболевания органов
ВВЕДЕНИЕ
Воспаление — это реакция иммунной системы на вредные стимулы, такие как патогены, поврежденные клетки, токсичные соединения или облучение [1], и действует, удаляя повреждающие раздражители и инициируя процесс заживления [2]. Таким образом, воспаление является защитным механизмом, жизненно важным для здоровья [3]. Обычно во время острых воспалительных реакций клеточные и молекулярные события и взаимодействия эффективно минимизируют надвигающуюся травму или инфекцию. Этот процесс смягчения способствует восстановлению гомеостаза тканей и разрешению острого воспаления. Однако неконтролируемое острое воспаление может стать хроническим, способствуя развитию множества хронических воспалительных заболеваний [4].
Таким образом, воспаление является защитным механизмом, жизненно важным для здоровья [3]. Обычно во время острых воспалительных реакций клеточные и молекулярные события и взаимодействия эффективно минимизируют надвигающуюся травму или инфекцию. Этот процесс смягчения способствует восстановлению гомеостаза тканей и разрешению острого воспаления. Однако неконтролируемое острое воспаление может стать хроническим, способствуя развитию множества хронических воспалительных заболеваний [4].
На тканевом уровне воспаление характеризуется покраснением, отеком, жаром, болью и потерей функции тканей, которые возникают в результате местных иммунных, сосудистых и воспалительных реакций клеток на инфекцию или травму [5].Важные микроциркуляторные события, которые происходят во время воспалительного процесса, включают изменения проницаемости сосудов, рекрутирование и накопление лейкоцитов, а также высвобождение медиатора воспаления [2, 6].
Различные патогенные факторы, такие как инфекция, повреждение тканей или инфаркт миокарда, могут вызывать воспаление, вызывая повреждение тканей. Этиология воспаления может быть инфекционной или неинфекционной (таблица). В ответ на повреждение ткани организм запускает каскад химических сигналов, который стимулирует реакции, направленные на исцеление пораженных тканей.Эти сигналы активируют хемотаксис лейкоцитов из общего кровотока к участкам повреждения. Эти активированные лейкоциты продуцируют цитокины, вызывающие воспалительные реакции [7].
Этиология воспаления может быть инфекционной или неинфекционной (таблица). В ответ на повреждение ткани организм запускает каскад химических сигналов, который стимулирует реакции, направленные на исцеление пораженных тканей.Эти сигналы активируют хемотаксис лейкоцитов из общего кровотока к участкам повреждения. Эти активированные лейкоциты продуцируют цитокины, вызывающие воспалительные реакции [7].
Таблица 1
Этиология воспаления
| Неинфекционные факторы | Инфекционные факторы |
|---|---|
| Физические факторы: ожоги, обморожения, телесные повреждения, инородные тела, травмы, ионизирующее излучение Химические вещества: глюкоза , жирные кислоты, токсины, алкоголь, химические раздражители (включая фторид, никель и другие микроэлементы) Биологическое: поврежденные клетки Психологическое: возбуждение | Бактерии вирусы другие микроорганизмы |
МЕХАНИЗМЫ ВОСПАЛИТЕЛЬНОЙ РЕАКЦИИ
Воспалительная реакция — это координируют активацию сигнальных путей, которые регулируют уровни медиатора воспаления в резидентных тканевых клетках и воспалительных клетках, рекрутируемых из крови [8].Воспаление является обычным патогенезом многих хронических заболеваний, включая сердечно-сосудистые заболевания и заболевания кишечника, диабет, артрит и рак [9]. Хотя процессы воспалительной реакции зависят от точной природы исходного стимула и его местоположения в организме, все они имеют общий механизм, который можно резюмировать следующим образом: 1) рецепторы структуры клеточной поверхности распознают вредные стимулы; 2) активируются воспалительные пути; 3) высвобождаются маркеры воспаления; и 4) рекрутируются воспалительные клетки.
Активация рецептора распознавания образов
Микробные структуры, известные как патоген-ассоциированные молекулярные паттерны (PAMP), могут запускать воспалительный ответ через активацию рецепторов распознавания паттернов (PRR), кодируемых зародышевой линией, экспрессируемых как в иммунных, так и в неиммунных клетках [10, 11] . Некоторые PRR также распознают различные эндогенные сигналы, активируемые при повреждении ткани или клетки, и известны как молекулярные паттерны, связанные с опасностями (DAMPS) [11]. DAMP являются биомолекулами хозяина, которые могут инициировать и поддерживать неинфекционный воспалительный ответ [12].Нарушенные клетки могут также рекрутировать врожденные воспалительные клетки в отсутствие патогенов, высвобождая DAMPs [13].
Классы семейств PRR включают Toll-подобные рецепторы (TLR), лектиновые рецепторы C-типа (CLR), рецепторы, индуцируемые ретиноевой кислотой (RIG), -I-подобные рецепторы (RLR) и NOD-подобные рецепторы (NLR). [5]. TLR представляют собой семейство высококонсервативных PRR млекопитающих, которые участвуют в активации воспалительного ответа [14]. Идентифицировано более десяти членов семейства TLR, и TLR являются наиболее хорошо изученными из известных PRR [15].Передача PAMP и DAMP опосредуется фактором дифференцировки миелоида-88 (MyD88) вместе с TLR. Передача сигналов через TLR активирует внутриклеточный сигнальный каскад [16, 17], который приводит к ядерной транслокации факторов транскрипции, таких как активаторный белок-1 (AP-1) и NF-κB или фактор регуляции интерферона 3 (IRF3) (рисунок). DAMPs и PAMPs имеют общие рецепторы, такие как TLR4, что указывает на сходство между инфекционными и неинфекционными воспалительными реакциями [18, 19].
Передача сигналов TLRMyD88-зависимые и TRIF-зависимые пути показаны.Передача сигналов через TLR активирует внутриклеточные сигнальные каскады, которые приводят к ядерной транслокации AP-1 и NF-κB или IRF3, которые регулируют воспалительный ответ.
Активация воспалительных путей
Воспалительные пути влияют на патогенез ряда хронических заболеваний и включают общие медиаторы воспаления и регулирующие пути. Воспалительные стимулы активируют внутриклеточные сигнальные пути, которые затем активируют выработку медиаторов воспаления. Первичные воспалительные стимулы, включая микробные продукты и цитокины, такие как интерлейкин-1β (IL-1β), интерлейкин-6 (IL-6) и фактор некроза опухоли-α (TNF-α), опосредуют воспаление через взаимодействие с TLR, IL -1 рецептор (IL-1R), рецептор IL-6 (IL-6R) и рецептор TNF (TNFR) [20].Активация рецептора запускает важные внутриклеточные сигнальные пути, включая митоген-активируемую протеинкиназу (MAPK), ядерный фактор каппа-B (NF-κB) и сигнальный преобразователь и активатор транскрипции (STAT) киназы Януса (JAK) [21–2] 23].
Путь NF-κB
Фактор транскрипции NF-κB играет важную роль в процессах воспаления, иммунного ответа, выживания и апоптоза [24]. Семейство NF-κB включает пять родственных факторов транскрипции: P50, p52, RelA (p65), RelB и c-Rel [25, 26].Активность NF-κB индуцируется рядом стимулов, включая патогенные вещества, межклеточные воспалительные цитокины и многие ферменты [27, 28]. В физиологических условиях белки IκB, присутствующие в цитоплазме, ингибируют NF-κB [29]. PRR используют аналогичные механизмы передачи сигнала для активации киназы IκB (IKK), которая состоит из двух субъединиц киназы, IKKα и IKKβ, и регуляторной субъединицы, такой как IKKγ. IKK регулирует активацию пути NF-κB посредством фосфорилирования IκB [8]. Фосфорилирование IκB приводит к его деградации протеасомой и последующему высвобождению NF-κB для ядерной транслокации и активации транскрипции генов [30].Этот путь регулирует выработку провоспалительных цитокинов и рекрутирование воспалительных клеток, которые способствуют воспалительной реакции (рисунок).
Путь NF-κBЭтот путь запускается TLR и воспалительными цитокинами, такими как TNF и IL-1, что приводит к активации комплексов RelA / p50, которые регулируют экспрессию воспалительных цитокинов. Для передачи сигналов NF-κB требуются субъединицы IKK. которые регулируют активацию пути посредством фосфорилирования IκB.
Путь MAPK
MAPK представляют собой семейство серин / треониновых протеинкиназ, которые направляют клеточные ответы на различные стимулы, включая осмотический стресс, митогены, тепловой шок и воспалительные цитокины (такие как IL-1, TNF-α и IL-6), которые регулируют пролиферацию, дифференцировку, выживаемость клеток и апоптоз [31, 32].MAPK млекопитающих включают регулируемую внеклеточными сигналами киназу ERK1 / 2, MAP-киназу p38 и N-концевые киназы c-Jun (JNK) [33]. Каждый путь передачи сигналов MAPK включает по меньшей мере три компонента: MAPK, киназу MAPK (MAPKK) и киназу киназы MAPK (MAPKKK). MAPKKKs фосфорилируют и активируют MAPKKs, которые, в свою очередь, фосфорилируют и активируют MAPKs [33, 34]. ERK обычно активируются митогенами и сигналами дифференцировки, тогда как воспалительные стимулы и стресс активируют JNK и p38 [35]. MKK1 и MKK2 активируют ERK1 / 2, MKK4 и MKK7 активируют JNK, а MKK3 и MKK6 активируют p38.Активация MAPK, включая Erk1 / 2, JNK, приводит к фосфорилированию и активации факторов транскрипции p38 v, присутствующих в цитоплазме или ядре, что инициирует воспалительный ответ [32, 36] (рисунок).
Путь MAPKЭтот путь опосредует внутриклеточную передачу сигналов, инициированную внеклеточными стимулами, такими как стресс и цитокины. MAPKKK фосфорилируют и активируют MAPKK, которые, в свою очередь, фосфорилируют и активируют MAPK. Семейство MAPK млекопитающих включает Erk1 / 2, JNK и p38.В пути Erk1 / 2 Erk1 / 2 активируется с помощью MKK1 / 2, который активируется с помощью Raf. В пути JNK JNK активируется с помощью MKK4 / 7, который активируется с помощью MEKK1 / 4, ASK1 и MLK3. В пути p38 p38 активируется с помощью MKK3 / 6, который активируется с помощью MLK3, TAK и DLK. Активированные MAPK фосфорилируют различные белки, включая факторы транскрипции, что приводит к регуляции воспалительных реакций.
Путь JAK-STAT
Высококонсервативный путь JAK-STAT включает различные цитокины, факторы роста, интерфероны и родственные молекулы, такие как лептин и гормон роста, и является сигнальным механизмом, с помощью которого внеклеточные факторы могут контролировать экспрессию генов [37 ].Связанные с рецептором JAK активируются лигандами и фосфорилируют друг друга, создавая сайты стыковки для STAT, которые являются латентными цитоплазматическими факторами транскрипции. Цитоплазматические STATs, рекрутируемые на эти сайты, подвергаются фосфорилированию и последующей димеризации перед транслокацией в ядро [38]. Фосфорилирование тирозина необходимо для димеризации STAT и связывания ДНК [39]. Следовательно, передача сигналов JAK / STAT обеспечивает прямую трансляцию внеклеточного сигнала в ответ транскрипции.Например, связывание членов семейства IL-6 с рецепторами плазматической мембраны активирует белки JAK-STAT. Белки STAT, перемещенные в ядро, связывают промоторные области гена-мишени для регуляции транскрипции воспалительных генов (рисунок) [40].
Путь JAK-STATПосле связывания IL-6 сигнал передается рецептором для активации JAK, которые затем активируют STAT. STAT дефосфорилируются в ядре, что приводит к активации нижележащих цитокинов.
Нарушение регуляции активности NF-κB, MAPK или JAK-STAT связано с воспалительными, аутоиммунными и метаболическими заболеваниями, а также раком [41].Передача сигналов через факторы транскрипции приводит к секреции цитокинов [42, 43]. Множественные факторы транскрипции регулируют множество воспалительных генов, таких как IL-1, TNF-α, IL-6 [44], колониестимулирующий фактор (CSF), интерфероны, трансформирующий фактор роста (TGF) и хемокины.
Воспалительные маркеры
Маркеры используются в клинических приложениях для обозначения нормальных биологических процессов по сравнению с патогенными и оценки реакции на терапевтические вмешательства. Маркеры воспаления могут быть предиктором воспалительных заболеваний [45–50] и коррелировать с причинами и последствиями различных воспалительных заболеваний, таких как сердечно-сосудистые заболевания, эндотелиальные дисфункции и инфекции [51, 52].Стимулы активируют воспалительные клетки, такие как макрофаги и адипоциты, и вызывают выработку воспалительных цитокинов, таких как IL-1β, IL-6, TNF-α, а также воспалительных белков и ферментов. Эти молекулы потенциально могут служить биомаркерами для диагностики заболеваний, прогноза и принятия терапевтических решений [53–57].
Воспалительные цитокины
Цитокины (таблица) преимущественно высвобождаются из иммунных клеток, включая моноциты, макрофаги и лимфоциты. Про- и противовоспалительные цитокины облегчают и подавляют воспаление соответственно.Воспалительные цитокины классифицируются как IL, колониестимулирующие факторы (CSF), IFN, TNF, TGF и хемокины, и продуцируются клетками, прежде всего, для рекрутирования лейкоцитов в место инфекции или повреждения [58]. Цитокины модулируют иммунный ответ на инфекцию или воспаление и регулируют само воспаление посредством сложной сети взаимодействий. Однако чрезмерная продукция воспалительных цитокинов может привести к повреждению тканей, гемодинамическим изменениям, органной недостаточности и, в конечном итоге, к смерти [59, 60]. Лучшее понимание того, как регулировать пути цитокинов, позволит более точно идентифицировать агент-опосредованное воспаление и лечить воспалительные заболевания [58].
Таблица 2
Резюме цитокинов и их функций
| Цитокин | Семья | Основные источники | Функция |
|---|---|---|---|
| IL-1β | IL-1 | Макрофаги, моноциты | Провоспалительный процесс, пролиферация, апоптоз, дифференцировка |
| IL-4 | IL-4 | Th-клетки | Противовоспалительное действие, пролиферация T-клеток и B-клеток, дифференцировка B-клеток |
| IL-6 | IL-6 | Макрофаги, Т-клетки, адипоциты | Провоспаление, дифференцировка, продукция цитокинов |
| IL-8 | CXC | Макрофаги, эпителиальные клетки, эндотелиальные клетки | Pro- воспаление, хемотаксис, ангиогенез |
| IL-10 | IL-10 | Моноциты, Т-клетки, B-клетки | Противовоспалительное действие, ингибирование провоспалительных цитокинов |
| IL-12 | IL-12 | Дендритные клетки, макрофаги, нейтрофилы | Провоспалительные процессы, дифференцировка клеток, активирует NK-клетки |
| IL-11 | IL-6 | Фибробласты, нейроны, эпителиальные клетки | Противовоспалительное действие, дифференцировка, индуцирует белок острой фазы |
| TNF-α | TNF | Макрофаги, NK-клетки, CD4 + лимфоциты, адипоциты | Провоспалительное, продукция цитокинов, пролиферация клеток, апоптоз, противоинфекция |
| IFN-γ | INF | Т-клетки, NK-клетки, NKT-клетки | Провоспалительный, врожденный, адаптивный иммунитет противовирусный |
| GM -CSF | IL-4 | Т-клетки, макрофаги, фибробласты | Провоспалительные процессы, активация макрофагов, увеличение функции нейтрофилов и моноцитов |
| T GF-β | TGF | Макрофаги, Т-клетки | Противовоспалительное, ингибирование продукции провоспалительных цитокинов |
Воспалительные белки и ферменты
Воспалительные белки в крови, включая С-реактивный белок (CRP) , гаптоглобин, сывороточный амилоид А, фибриноген и альфа-1-кислотный гликопротеин [61] помогают восстановить гомеостаз и уменьшить рост микробов независимо от антител во время травмы, стресса или инфекции [62].Аномальная активация определенных ферментов, включая высокоподвижный блок 1 группы (HMGB1), супероксиддисмутазу (SOD), глутатионпероксидазу (GPx), NADPH-оксидазу (NOX), индуцибельную синтазу оксида азота (iNOS) и циклооксигеназу (COX) -2, играют ключевую роль в развитии заболеваний, связанных с воспалением, таких как сердечно-сосудистые заболевания и рак [63–66]. Например, внеклеточные эффекты HMGB1 могут быть опосредованы активацией связанных с TLR сигнальных путей [67]. Первичной мишенью внеклеточного HMGB1 является TLR4 [68], который запускает MyD88-зависимые внутриклеточные сигнальные каскады, участвующие в активации путей NF-κB и MAPK.Это приводит к высвобождению таких воспалительных цитокинов, как TNF-α и IL-1β [67]. Воспалительные белки и ферменты используются в медицине в качестве биомаркеров воспалений, инфекций и травм.
Другие маркеры воспаления
Системы антиоксидантной защиты, включая антиоксидантные ферменты, влияют на окислительный стресс. Повышенный окислительный стресс может вызывать продукцию активных форм кислорода (ROS), малонового диальдегида (MDA), 8-гидрокси-2-дезоксигуанозина (8-OHdG) и изопростанов [64, 69], каждый из которых может активировать различные факторы транскрипции, включая NF -κB, AP-1, p53 и STAT.Таким образом, этот каскад может увеличивать экспрессию генов, кодирующих факторы роста, воспалительные цитокины и хемокины [70]. Окислительный стресс связан с патогенезом множества заболеваний, таких как сердечно-сосудистые заболевания, рак, диабет, гипертония, старение и атеросклероз. Следовательно, продукты окислительного стресса также могут использоваться в качестве маркеров воспалительной реакции.
Типы клеток при воспалительной реакции
Воспалительная реакция включает в себя хорошо скоординированную сеть из многих типов клеток.Активированные макрофаги, моноциты и другие клетки опосредуют местные реакции на повреждение ткани и инфекцию. В местах повреждения ткани поврежденные эпителиальные и эндотелиальные клетки высвобождают факторы, запускающие воспалительный каскад, наряду с хемокинами и факторами роста, которые привлекают нейтрофилы и моноциты. Первыми клетками, привлекаемыми к месту повреждения, являются нейтрофилы, за ними следуют моноциты, лимфоциты (естественные клетки-киллеры [NK-клетки], Т-клетки и В-клетки) и тучные клетки [71–73]. Моноциты могут дифференцироваться в макрофаги и дендритные клетки и рекрутируются посредством хемотаксиса в поврежденные ткани.Опосредованные воспалением изменения иммунных клеток связаны со многими заболеваниями, включая астму, рак, хронические воспалительные заболевания, атеросклероз, диабет, а также аутоиммунные и дегенеративные заболевания.
Нейтрофилы, которые нацелены на микроорганизмы в организме, также могут повреждать клетки и ткани хозяина [74]. Нейтрофилы являются ключевыми медиаторами воспалительного ответа и программируют антигенпрезентирующие клетки для активации Т-клеток и высвобождения локализованных факторов для привлечения моноцитов и дендритных клеток [7].Макрофаги являются важными компонентами системы мононуклеарных фагоцитов и имеют решающее значение для инициации, поддержания и разрешения воспаления [75]. Во время воспаления макрофаги представляют антигены, подвергаются фагоцитозу и модулируют иммунный ответ, продуцируя цитокины и факторы роста. Тучные клетки, которые находятся в матрицах соединительной ткани и на эпителиальных поверхностях, являются эффекторными клетками, которые инициируют воспалительные реакции. Активированные тучные клетки высвобождают множество медиаторов воспаления, включая цитокины, хемокины, гистамин, протеазы, простагландины, лейкотриены и протеогликаны серглицина [76].
Несколько групп продемонстрировали, что тромбоциты влияют на воспалительные процессы, от атеросклероза до инфекции. Взаимодействие тромбоцитов с воспалительными клетками может опосредовать провоспалительные исходы. Ответ острой фазы (APR) — это самый ранний ответ на инфекцию или травму, и некоторые исследования показали, что тромбоциты индуцируют APR [77]. Будучи задействованными воспалительными стимулами, иммунные клетки усиливают и поддерживают APR, высвобождая местные медиаторы воспаления в месте набора.
РАЗРЕШЕНИЕ ВОСПАЛЕНИЯ
Чтобы предотвратить прогрессирование острого воспаления в стойкое хроническое воспаление, воспалительная реакция должна быть подавлена, чтобы предотвратить дополнительное повреждение тканей. Разрешение воспаления — это хорошо управляемый процесс, включающий производство медиаторов, контролируемое пространственно и во времени, во время которого градиенты хемокинов со временем растворяются. Циркулирующие белые кровяные тельца в конечном итоге больше не воспринимают эти градиенты и не рекрутируются в места повреждения.Нарушение регуляции этого процесса может привести к неконтролируемому хроническому воспалению [78]. Процессы разрешения воспаления, которые исправляют гомеостаз ткани, включают уменьшение или прекращение инфильтрации ткани нейтрофилами и апоптоз отработанных нейтрофилов, противорегуляцию хемокинов и цитокинов, трансформацию макрофагов из классически активированных клеток в альтернативно активированные и начало заживления [79, 80].
Хроническое воспаление возникает, когда острые воспалительные механизмы не в состоянии устранить повреждение тканей [81], и может привести к множеству заболеваний, таких как сердечно-сосудистые заболевания, атеросклероз, диабет 2 типа, ревматоидный артрит и рак [82].Понимание общих механизмов, которые управляют дисфункцией в различных системах органов, позволит разрабатывать и производить улучшенные целевые методы лечения.
ОРГАН-СПЕЦИАЛЬНЫЕ ВОСПАЛИТЕЛЬНЫЕ РЕАКЦИИ
Воспаление уже давно признано основной причиной заболеваний. По оценкам, около 15% случаев рака у человека связаны с хронической инфекцией и воспалением [83]. Острое и хроническое повреждение тканей, опосредованное воспалением, наблюдается во многих системах органов, включая сердце, поджелудочную железу, печень, почки, легкие, мозг, кишечник и репродуктивную систему.
Сердце
Сердечно-сосудистые заболевания и лежащая в их основе патология, атеросклероз, являются основной причиной смерти и инвалидности во всем мире [84, 85]. По прогнозам, к 2030 году от сердечно-сосудистых заболеваний ежегодно будет умирать почти 23,6 миллиона человек [86, 87]. Медиаторы воспаления играют ключевую роль в развитии атеросклероза, от начального набора лейкоцитов до разрыва атеросклеротической бляшки [88–91]. Воспаление также является ранним событием сердечного стресса. В пораженных сердечных тканях наблюдаются повышенные уровни молекул адгезии эндотелия и повышенная продукция и высвобождение воспалительных цитокинов и хемокинов [92].
Врожденная иммунная система является основной защитой сердца от патогенов и повреждений тканей [93]. Инфаркт миокарда, который обычно возникает в результате коронарного атеросклероза и включает острую потерю многих клеток миокарда, является наиболее частой причиной повреждения сердца [94]. Некротические сердечные клетки инициируют воспалительный каскад, чтобы удалить мертвые клетки и мусор из инфаркта [95, 96]. Смерть клетки высвобождает внутриклеточные компоненты, которые активируют механизмы врожденного иммунитета, чтобы вызвать воспалительную реакцию.Эндогенные лиганды, высвобождаемые после повреждения, распознаются рецепторами клеточной поверхности как сигналы опасности и активируют воспаление [97, 98]. TLR-опосредованные пути запускают постинфарктные воспалительные реакции путем активации передачи сигналов NF-κB [98–103]. Хемокины привлекают воспалительные лейкоциты к инфаркту, а цитокины способствуют адгезии лейкоцитов к эндотелиальным клеткам [104, 105]. Более того, TGF-β и IL-10 способствуют восстановлению сердца, подавляя воспаление, усиливая модуляцию фенотипа миофибробластов и способствуя отложению внеклеточного матрикса [106, 107].
Сердечно-сосудистые заболевания являются основной причиной смерти и инвалидности у пациентов с сахарным диабетом, особенно у пациентов с диабетом 2 типа (СД2), у которых сердечно-сосудистые заболевания возникают в среднем на 14,6 лет раньше [108]. Около двух третей смертей людей с диабетом вызваны сердечно-сосудистыми заболеваниями; среди них примерно 40% умирают от ишемической болезни сердца, 15% — от других форм сердечных заболеваний, в основном от застойной сердечной недостаточности и около 10% — от инсульта [109]. Недавние глобальные оценки показывают, что более 422 миллионов взрослых в настоящее время живут с диабетом, из которых более 90% страдают СД2.
Диабет — это группа метаболических нарушений, характеризующихся устойчивым высоким уровнем сахара в крови, и представляет собой серьезную глобальную проблему для здоровья как отдельных людей и их семей, так и систем здравоохранения [110]. Осложнения диабета включают сердечный приступ, инсульт, почечную недостаточность, ампутацию конечностей, слепоту и повреждение нервов. Диабет возникает либо из-за нарушения выработки инсулина в поджелудочной железе, либо из-за того, что клетки организма не реагируют на продуцируемый инсулин [111]. Инсулинорезистентность определяется как пониженное потребление глюкозы, стимулированное инсулином, и связано с малоподвижностью, ожирением и старением.Клетки островков поджелудочной железы реагируют на инсулинорезистентность увеличением секреции инсулина и клеточной массы. Однако, когда островковые β-клетки неспособны компенсировать инсулинорезистентность, развивается дефицит инсулина, за которым следует СД2 [112], который все чаще характеризуется как воспалительное заболевание [113, 114]. Повышенные уровни циркулирующих белков острой фазы, включая CRP, фибриноген, сывороточный амилоид A, ингибитор активатора плазминогена и гаптоглобин, а также сиаловую кислоту, цитокины и хемокины, наблюдались у пациентов с T2D.Повышенные уровни IL-1β, IL-6, TNF-α и CRP также являются предиктором СД2. Уровень антагониста рецептора IL-1 (IL-1RA) повышен при ожирении и преддиабете до начала T2D. Чрезмерный уровень питательных веществ, в том числе глюкозы и свободных жирных кислот, способствует инсулинорезистентности. T2D также активирует пути NF-κB, MAPK и JAK-STAT, каждый из которых может способствовать воспалению тканей [110, 114, 115]. Метаболические стрессоры также негативно влияют на клетки островков поджелудочной железы и чувствительные к инсулину ткани, включая жировую ткань, способствуя выработке и высвобождению местных цитокинов и хемокинов.В то же время задействуются иммунные клетки, такие как тучные клетки и макрофаги, которые способствуют воспалению тканей. Точно так же высвобождение цитокинов и хемокинов из жировой ткани в кровоток способствует дальнейшему воспалению в других тканях [116].
Поджелудочная железа
Панкреатит, вызванный обструкцией протока поджелудочной железы, мутацией гена трипсиногена или алкоголизмом, является воспалительным заболеванием поджелудочной железы [117]. Заболеваемость острым панкреатитом (ОП) колеблется от 4 до 45 на 100 000 пациентов в год и ежегодно увеличивается примерно на 1.3–4,0% в большинстве развитых стран. АП является одной из наиболее частых желудочно-кишечных причин госпитализации в США, а хронический панкреатит (ХП) встречается реже, чем АП. Однако пациенты с ХП испытывают хроническую боль в животе и внешнесекреторную и / или эндокринную недостаточность, что приводит к снижению качества жизни [118]. Панкреатит характеризуется деструкцией ацинарных клеток и активацией воспалительных клеток, включая макрофаги, нейтрофилы и гранулоциты, которые секретируют воспалительные цитокины [117, 119].Эти цитокины дополнительно активируют звездчатые клетки поджелудочной железы (PSC), чтобы способствовать CP [120]. Для развития панкреатита необходимы различные молекулярные пути, такие как NF-κB, MAPK и JAK-STAT, которые играют критическую роль в активации воспалительных клеток при панкреатите [117].
Рак поджелудочной железы (ПК) остается одним из самых смертоносных злокачественных новообразований и серьезным бременем для здоровья [121], а также четвертой по частоте причиной смерти от рака в США [118]. Существует сильная связь между предшествующим CP и PC [122].ХП приводит к фиброзу, который является частым патологическим признаком и основным фактором риска ПК [123]. Рак поджелудочной железы возникает в результате нарушения регуляции онкогенов и генов-супрессоров опухолей, а также факторов роста и их рецепторов, включая эпидермальные факторы роста, фактор роста эндотелия сосудов (VEGF), фактор роста фибробластов (FGF) и многие цитокины, такие как TGF-β. , IL-1, IL-6, TNF-α и IL-8, которые модулируют пути, участвующие в росте и дифференцировке [124, 125]. Ши, и др. показал, что VEGF активируется низкой внеклеточной PH (ацидозом), который часто возникает вокруг некротических областей в опухолях, и что ацидоз активирует IL-8 [126].VEGF и IL-8 являются важными ангиогенными факторами при PC [126], а активация этих генов, способствующая ацидозу, может быть опосредована трансактивацией и кооперацией NF-κB и AP-1 [127].
Печень
Воспаление в печени защищает этот орган от инфекции и травм, но чрезмерное воспаление может привести к значительной потере гепатоцитов, ишемии-реперфузии, метаболическим изменениям и, в конечном итоге, необратимому повреждению печени [128]. Воспаление может разрушить клетки паренхимы печени, увеличивая риск хронических заболеваний печени, таких как неалкогольная жировая болезнь печени (НАЖБП) или вирусный гепатит.Хронические заболевания печени — основная причина заболеваемости и смертности в США [129].
Печень — самый большой твердый орган в организме [130], являющийся мишенью как инфекционных, так и неинфекционных воспалительных патологий. Инфекционное воспаление печени в основном вызывается микроорганизмами, такими как бактериальные продукты, вирус гепатита B (HBV) или вирус гепатита C (HCV) [131, 132]. Стерильное воспаление (SI) также играет важную роль в патологии многих заболеваний печени, таких как алкогольный или неалкогольный стеатогепатит, лекарственное поражение печени и ишемия / реперфузия [133–135].В SI эндогенные DAMPs высвобождаются в поврежденные ткани и активируют иммунные клетки [136]. Хотя воспаление, вызванное патогенами, и SI различаются, они имеют несколько общих функциональных характеристик. Многие рецепторы и пути могут быть активированы как PAMP, так и DAMP [137]. TLR4, например, может быть активирован бактериальным LPS и клеточным HMGB1. Благодаря уникальному кровоснабжению печени, PAMP кишечного происхождения и DAMP из гепатоцитов способствуют воспалению при различных заболеваниях. Например, активация PRR с помощью DAMP и PAMP может индуцировать продукцию провоспалительных цитокинов и локализацию иммунных клеток в местах повреждения.Распознавание DAMP и PAMP приводит к сборке инфламмасомы, комплекса цитозольных белков, который активирует сериновую протеазу каспазу-1 с последующей активацией и секрецией IL-1β и других цитокинов. В то же время активация клеток Купфера и привлечение воспалительных клеток приводит к выработке цитокинов и хемокинов, которые способствуют долгосрочному воспалению, повреждению гепатоцитов и / или холестазу [138].
Легкое
Воспалительные заболевания легких включают сложные взаимодействия между структурными и иммунными клетками [139].Воспаление легких возникает преимущественно в результате воздействия на ткани бактериальных и вирусных патогенов и / или загрязнителей окружающей среды. Чрезмерное острое воспаление и последующее повреждение легких могут вызвать фиброз легких и нарушить газообмен. Неразрешенное повреждение легких и хроническое воспаление часто наблюдаются при остром респираторном дистресс-синдроме, муковисцидозе, хронической обструктивной болезни легких (ХОБЛ) и астме [140–142]. Примерно 90% случаев ХОБЛ связаны с вызванным курением сигаретным воспалением в мелких дыхательных путях и паренхиме легких [143].Курение сигарет является основным фактором риска развития ХОБЛ, которая включает как системное, так и легочное воспаление. Длительное курение может вызвать инфильтрацию макрофагов, нейтрофилов и активированных Т-лимфоцитов в дыхательные пути и способствовать выработке хемокинов, кислородных радикалов, протеаз и цитокинов, включая TNF-α, IL-6 и IL-8, в легких. [144].
Почки
Воспаление почек способствует прогрессирующему повреждению почек, которое может привести к гломерулонефриту, терминальной стадии почечной недостаточности или острой или хронической болезни почек (ХБП) [145–147].Примерно 10–12% населения страдает ХБП, и примерно у 50% пожилых пациентов наблюдаются признаки дисфункции почек, что связано с высокой заболеваемостью и смертностью [52]. Воспаление почек чаще всего вызывается инфекцией, ишемией / реперфузией, образованием / отложением иммунных комплексов in situ или нарушением регуляции пути комплемента [145]. ХБП и острое повреждение почек (ОПП) являются наиболее тяжелыми типами заболеваний почек [148]. Интерстициальное воспаление и повреждение канальцев обычно наблюдаются при остром и хроническом повреждении почек.Эпителиальные клетки почечных канальцев, вероятно, являются важными промоторами воспаления почек, секретируя различные воспалительные цитокины в ответ как на иммунные, так и на неиммунные факторы, а инфильтрация лейкоцитов зависит от местного присутствия этих цитокинов [146]. Стимулы, которые могут вызвать повреждение почек, активируют факторы транскрипции (NF-κB или MAPK). Эти стимулы включают цитокины, факторы роста, DAMP и PAMP, TLR, Nod-подобные рецепторы (NLR), а также метаболические (высокое содержание глюкозы, конечные продукты гликозилирования) и иммунные медиаторы [147].
Кишечник
Острые и хронические воспалительные заболевания кишечника могут вызывать различные проблемы со здоровьем и снижать качество жизни пациентов во всем мире [149, 150]. Сложные полигенетические воспалительные заболевания кишечника (ВЗК) характеризуются чрезмерной воспалительной реакцией на микробную флору кишечника [151]. ВЗК в основном включают язвенный колит (ЯК) и болезнь Крона (БК), а также неинфекционное воспаление кишечника [152, 153]. Идиопатические ВЗК, такие как БК и ЯК, вызываются цитокиновым неинфекционным воспалением кишечника.Например, CD ассоциирован с избыточным продуцированием IFN-γ / IL-17 и IL-12 / IL-23, тогда как UC связан с избытком IL-13 [153]. Таким образом, ВЗК, по-видимому, является результатом дисфункционального взаимодействия между кишечными бактериями и иммунной системой слизистой оболочки [154]. Ключевым процессом в ответе иммунной системы на микробы является распознавание микробных агентов через PRR, включая TLR и нуклеотид-связывающий домен олигомеризации, содержащий NLR, которые распознают эволюционно консервативные PAMP [155]. При обнаружении PAMP PRR активируют внутриклеточные сигнальные пути, которые индуцируют выработку цитокинов и хемокинов, способствующих устойчивости хозяина к инфекции.Передача сигналов TLR (в основном TLR4) индуцирует транскрипцию NF-κB и MAPK. В то же время NLR также активируются посредством обнаружения лиганда [154], в свою очередь, активируя каспазу-1 с последующей активацией и секрецией IL-1β, интерлейкина-18 и других цитокинов [154].
Репродуктивная система
Признаки воспаления наблюдаются во время многих нормальных репродуктивных процессов, включая менструацию, овуляцию, имплантацию и роды [156]. Травма и заживление, вызванные менструацией, овуляцией и родами, запускают воспалительный каскад.Однако инициирование и поддержание воспалительных процессов также являются важными компонентами многих заболеваний репродуктивного тракта. Поврежденные ткани локально выделяют воспалительные интерлейкины, факторы роста, цитокины и простагландины, которые активируют сигнальные пути и привлекают иммунные клетки (например, нейтрофилы и макрофаги) к месту повреждения. Этот процесс синергетически контролирует ремоделирование и восстановление тканей, но также может вызывать воспалительные заболевания [7]. Воспалительные цитокины, включая IL-6, являются основными медиаторами заболеваний репродуктивного тракта, связанных с воспалением, и действуют через пути передачи сигналов, такие как путь MAPK [157, 158].
Мозг
Воспалительные реакции мозга возникают при многих заболеваниях центральной нервной системы (ЦНС), включая аутоиммунные заболевания, нейродегенеративные заболевания, такие как болезнь Альцгеймера (БА) и болезнь Паркинсона (БП), а также эпилепсия. Воспалительные реакции в головном мозге могут повышать возбудимость нейронов, повреждать клетки и увеличивать проницаемость гематоэнцефалического барьера для различных молекул [159–161]. Связанные с воспалением заболевания ЦНС возникают в результате активации резидентных иммунных клеток головного мозга и микроглии, которые продуцируют провоспалительные маркеры [162].Эти воспалительные процессы также затрагивают как врожденную, так и адаптивную иммунные системы и напоминают иммунные реакции на системную инфекцию. Цитокины и TLR являются основными медиаторами воспаления при переходе от врожденного к адаптивному. Воспалительные реакции в ЦНС также могут быть вызваны эндогенными лигандами, распознаваемыми TLR. DAMP, такие как белки теплового шока и молекулы деградации внеклеточного матрикса, проникая в мозг через поврежденный гематоэнцефалический барьер, могут инициировать воспалительные реакции.Воспалительная реакция ЦНС является сильной как реакция на инфекционные агенты, так и на повреждение головного мозга, такое как повреждение тканей, наблюдаемое после ишемического, травматического или эксайтотоксического повреждения головного мозга или судорог [160, 163, 164].
Что это такое, причины, симптомы и лечение
Обзор
Что такое воспаление?
Когда ваше тело сталкивается с возбудителем (например, вирусами, бактериями или токсичными химическими веществами) или получает травму, оно активирует вашу иммунную систему. Ваша иммунная система высылает свои первые реагенты: воспалительные клетки и цитокины (вещества, которые стимулируют больше воспалительных клеток).
Эти клетки начинают воспалительную реакцию, улавливая бактерии и другие возбудители болезней, или начинают заживлять поврежденную ткань. Результатом может стать боль, отек, синяк или покраснение. Но воспаление также влияет на невидимые системы организма.
В чем разница между острым воспалением и хроническим воспалением?
Есть два типа воспаления:
- Острое воспаление: Реакция на внезапное повреждение тела, например, порезание пальца.Чтобы залечить порез, ваше тело отправляет воспалительные клетки в травму. Эти клетки запускают процесс заживления.
- Хроническое воспаление: Ваше тело продолжает посылать воспалительные клетки, даже когда нет внешней опасности. Например, при ревматоидном артрите воспалительные клетки и вещества атакуют ткани суставов, что приводит к воспалению, которое возникает и уходит, и может вызывать серьезные повреждения суставов с болью и деформациями.
Каковы симптомы острого и хронического воспаления?
Острое воспаление может вызвать:
- Покраснение кожи в месте травмы.
- Боль или нежность.
- Отек.
- Тепло.
Симптомы хронического воспаления обнаружить труднее, чем симптомы острого воспаления. Признаки хронического воспаления могут включать:
Какие состояния связаны с хроническим воспалением?
Хроническое воспаление участвует в развитии многих состояний, в том числе:
Возможные причины
Каковы наиболее частые причины воспалений?
К наиболее частым причинам хронического воспаления относятся:
- Аутоиммунные заболевания , например волчанка, при которой ваше тело атакует здоровые ткани.
- Воздействие токсинов, загрязнений или промышленных химикатов.
- Нелеченное острое воспаление , например, в результате инфекции или травмы.
Некоторые факторы образа жизни также способствуют воспалительным процессам в организме. Вероятность развития хронического воспаления выше, если вы:
- Чрезмерное употребление алкоголя.
- Иметь высокий индекс массы тела (ИМТ), который соответствует диапазону ожирения, если только он не является очень мускулистым.
- Выполняйте упражнения с максимальной интенсивностью слишком часто или вы недостаточно тренируетесь.
- Испытывает хронический стресс.
- Дым.
Уход и лечение
Как лечить воспаление?
Воспаление не всегда требует лечения. При остром воспалении покой, лед и хороший уход за раной часто снимают дискомфорт за несколько дней.
Если у вас хроническое воспаление, ваш лечащий врач может порекомендовать:
- Добавки : Некоторые витамины (витамин A, витамин C, витамин D) и добавки (цинк) могут уменьшить воспаление и улучшить восстановление.Например, ваш лечащий врач может прописать добавку с рыбьим жиром или витамин (ы). Или вы можете использовать специи с противовоспалительными свойствами, такие как куркума, имбирь или чеснок.
- Нестероидные противовоспалительные препараты (НПВП) : Эти лекарства, отпускаемые без рецепта, уменьшают воспаление. Ваш лечащий врач может порекомендовать ибупрофен (Advil®), аспирин (Bayer®) или напроксен (Aleve®).
- Инъекции стероидов : Уколы кортикостероидов уменьшают воспаление в определенном суставе или мышце.Например, если у вас ревматоидный артрит, поражающий вашу спину, ваш лечащий врач может сделать укол стероидов в позвоночник. Вам не следует делать более трех-четырех инъекций стероидов в одну и ту же часть тела в год.
Что я могу сделать дома для лечения воспаления?
Вы можете выбрать противовоспалительную диету. Некоторые исследования показывают, что у людей, которые придерживаются средиземноморской диеты, меньше воспалений в организме.
Вы можете съесть больше продуктов с противовоспалительными свойствами, например:
- Жирная рыба, например скумбрия, лосось или сардины.
- Листовая зелень, например шпинат и капуста.
- Оливковое масло.
- Помидоры.
Слишком большое количество определенных продуктов может усилить воспаление. Если у вас хроническое воспаление, вы можете почувствовать себя лучше, если не будете принимать:
- Жареные продукты, в том числе многие продукты быстрого приготовления.
- Колбасные изделия с нитратами, например хот-доги.
- Масла высокой степени очистки и трансжиры.
- Рафинированные углеводы, например сахар, выпечка или белый хлеб.
Как предотвратить воспаление?
Вы можете снизить риск хронического воспаления, выработав здоровый образ жизни.Вот некоторые из этих привычек:
Когда звонить доктору
Когда мне обращаться к врачу по поводу воспаления?
Обратитесь к своему врачу, если вы получили серьезную травму. Также поговорите со своим врачом, если у вас постоянная боль, отек, скованность или другие симптомы. Эксперт в области здравоохранения может сузить круг причин и найти способы помочь вам почувствовать себя лучше.
Записка из клиники Кливленда
Воспаление — важная часть процесса заживления вашего тела.Это происходит, когда воспалительные клетки перемещаются к месту травмы или инородного тела, такого как бактерии. Если воспалительные клетки остаются слишком долго, это может привести к хроническому воспалению. Хроническое воспаление является симптомом других заболеваний, например ревматоидного артрита. Ваш лечащий врач может порекомендовать лекарства или лечение на дому. Вы можете уменьшить воспаление, употребляя противовоспалительные продукты и управляя стрессом.
Что такое воспаление? — InformedHealth.org
Если рана опухает, краснеет и болит, это может быть признаком воспаления.Вообще говоря, воспаление — это реакция иммунной системы организма на раздражитель. Раздражителем может быть микроб, но также может быть посторонний предмет, например, заноза в пальце.
Это означает, что воспаление начинается не только тогда, когда, например, рана уже инфицирована бактериями, сочится гноем или плохо заживает. Он начинается уже тогда, когда организм пытается бороться с вредным раздражителем.
Причины воспаления
Воспаления могут вызывать разные вещи.Это наиболее распространенные:
- Патогены (микробы), такие как бактерии, вирусы или грибки
-
Внешние травмы, такие как царапины или повреждения посторонними предметами (например, шипом в пальце)
-
Воздействие химикатов или радиации
Заболевания или медицинские состояния, вызывающие воспаление, часто имеют название, оканчивающееся на «-ит». Например:
-
Цистит: воспаление мочевого пузыря
- Бронхит: воспаление бронхов
-
Отит: воспаление среднего уха
-
Дерматит: заболевание, при котором воспаляется кожа
Признаки воспаления
Есть пять симптомов, которые могут быть признаками острого воспаления:
-
Покраснение
-
Тепло
-
Отек
-
Боль 85
90ss
Примеры потери функции включают невозможность двигать воспаленным суставом должным образом, ухудшение обоняния во время простуды или затруднение дыхания при бронхите.
Воспаления не всегда вызывают все пять симптомов. Некоторые воспаления протекают «незаметно» и не вызывают никаких симптомов.
Общие реакции в организме
Если воспаление сильное, оно может вызвать общие реакции в организме. Они могут включать следующие признаки и симптомы:
-
Общее недомогание, истощение и жар. Это признаки того, что иммунная система очень активна и нуждается в большом количестве энергии, которой может не хватать для других видов деятельности.Если скорость метаболизма выше из-за лихорадки, может производиться больше антител и клеток иммунной системы.
-
Изменения в крови, например увеличение количества клеток иммунной системы.
Очень редкое, но опасное осложнение инфекции называется септицемией (заражением крови). Возможные признаки этого осложнения включают озноб, очень плохое самочувствие и очень высокую температуру.
Септицемия может возникнуть, если бактерии быстро размножаются в определенной части тела, а затем внезапно многие из них попадают в кровоток.Это может произойти, если организм не может локально бороться с инфекцией, если микробы очень агрессивны или если иммунная система очень слаба. Септицемия требует неотложной медицинской помощи и требует как можно скорее лечения у врача.
Что происходит, когда у вас воспаление
Когда в вашем теле возникает воспаление, может быть задействовано множество различных клеток иммунной системы. Они выделяют различные вещества, известные как медиаторы воспаления. К ним относятся гормоны брадикинин и гистамин.Они заставляют мелкие кровеносные сосуды в ткани расширяться (расширяться), позволяя большему количеству крови достигать поврежденной ткани. По этой причине воспаленные участки краснеют и становятся горячими.
Увеличенный кровоток также позволяет переносить большее количество клеток иммунной системы к поврежденной ткани, где они помогают в процессе заживления. Более того, оба эти гормона раздражают нервы и вызывают передачу болевых сигналов в мозг. Это имеет защитную функцию: если воспаление болит, вы, как правило, защищаете пораженную часть тела.
Медиаторы воспаления выполняют еще одну функцию: они облегчают выход клеткам иммунной системы из мелких кровеносных сосудов, чтобы большее их количество могло попасть в пораженные ткани. Клетки иммунной системы также заставляют больше жидкости попадать в воспаленную ткань, из-за чего она часто опухает. Отек снова спадает через некоторое время, когда эта жидкость выводится из тканей.
Слизистые оболочки также выделяют больше жидкости при воспалении. Например, это происходит, когда у вас заложен нос и воспаляются слизистые оболочки носа.Тогда дополнительная жидкость поможет быстро вывести вирусы из вашего тела.
Воспаления тоже могут вызывать хронические заболевания.
Воспаления не всегда помогают организму. При некоторых заболеваниях иммунная система по ошибке борется с собственными клетками организма, вызывая опасные воспаления. К ним относятся, например:
-
Ревматоидный артрит, при котором постоянно воспалены многие суставы по всему телу
-
Псориаз — хроническое кожное заболевание
В совокупности известные как хронические воспалительные заболевания, эти заболевания могут длиться до годы или даже всю жизнь.Их выраженность и уровень активности различны.
Источники
-
Andreae S. Lexikon der Krankheiten und Untersuchungen. Штутгарт: Тиме; 2008.
-
Касперс Д.Л., Хаузер С.Л., Джеймсон Дж.Л., Фаучи А.С., Лонго Д.Л., Лоскальцо Дж. Харрисон. Принципы внутренней медицины. Нью-Йорк: компании McGraw-Hill. 19 изд; 2015.
-
Pschyrembel W. Klinisches Wörterbuch. Берлин: Де Грюйтер; 2017.
-
Информация о здоровье IQWiG написана с целью помочь люди понимают преимущества и недостатки основных вариантов лечения и здоровья услуги по уходу.
Поскольку IQWiG — немецкий институт, некоторая информация, представленная здесь, относится к Немецкая система здравоохранения. Пригодность любого из описанных вариантов в индивидуальном случай можно определить, посоветовавшись с врачом. Мы не предлагаем индивидуальных консультаций.
Наша информация основана на результатах качественных исследований. Это написано команда медицинские работники, ученые и редакторы, а также рецензируются внешними экспертами. Ты сможешь найти подробное описание того, как создается и обновляется наша медицинская информация в наши методы.
Воспаление мягких тканей — обзор
Лабораторные исследования
Не существует единого лабораторного теста, который бы отличал ЮИА от других заболеваний. Основная роль лабораторных исследований — исключить другие заболевания, например лейкоз или инфекционный артрит. Антинуклеарные антитела (ANA), ревматоидный фактор (RF) и HLA-B27 не обладают какой-либо дискриминационной способностью идентифицировать детей с ЮИА. В основном они полезны для дальнейшей категоризации и прогнозирования результата.
Скорость оседания эритроцитов (СОЭ), уровень С-реактивного белка (СРБ) и количество тромбоцитов — полезные тесты для дифференциации детей с ЮИА от детей с невоспалительными заболеваниями. Однако нормальный уровень СОЭ и / или СРБ нельзя использовать для исключения ЮИА при наличии убедительных клинических признаков, потому что почти половина детей с ЮИА имеют нормальные значения в начале заболевания в популяционных условиях. 7 Уровни СОЭ и СРБ могут быть очень высокими при системных и полиартикулярных категориях ЮИА, также может присутствовать тромбоцитоз.
Анемия может возникать при всех формах ЮИА, но более выражена в системных и полиартикулярных группах. Обычно анемия является вторичной по отношению к хроническому воспалению, что отражается низким уровнем сывороточного железа, низкой железосвязывающей способностью и нормальными запасами гемосидерина. Уровень ферритина в сыворотке крови может быть повышен, потому что это белок острой фазы, отражающий воспалительную активность, а не запасы железа. При системном ЮИА могут быть обнаружены очень высокие уровни ферритина, которые являются сигналом об осложнении синдрома активации макрофагов. 8 Лечение кортикостероидами и нестероидными противовоспалительными препаратами может вызвать потерю крови в кишечнике и вторичный дефицит железа, который обычно поддается лечению добавками железа.
Лейкоцитоз с преобладанием нейтрофилов наблюдается у детей с системным ЮИА, а также часто встречается у детей с полиартикулярным ЮИА. Хотя легкий или умеренный тромбоцитоз является обычным явлением, тромбоцитопения не является частью ЮИА и требует обследования для выявления системной красной волчанки (СКВ) или злокачественных новообразований, особенно лейкемии.Уровни сывороточного иммуноглобулина могут быть повышены, как и титры антистрептококков, даже при отсутствии недавней стрептококковой инфекции.
АНА обнаруживаются в 40–85% случаев олигоартикулярного ЮИА с ранним началом и связаны с хроническим увеитом. Специфический антинуклеарный антиген (ы), ответственный за положительность ANA при ЮИА, не идентифицирован. Тем не менее, есть некоторые признаки того, что антигистоны могут быть связаны с ЮИА и служить предикторами увеита. 9 ANA, специфичные для заболевания, которые выявляются с помощью большинства тестов ANA, основанных на иммуноферментном анализе, не связаны с ЮИА или увеитом, и, если они обнаружены, требуется обследование на СКВ у подростков, особенно у подростков с артритом.
Иммуноглобулин M (IgM) RF и антитела к цитруллинированному белку (ACPA) почти исключительно присутствуют у подростков с полиартикулярным ЮИА. 10 Иммунофлуоресценция ANA и RF IgM могут временно и неспецифически присутствовать при любой воспалительной реакции. Для того чтобы эти маркеры были распознаны как дескрипторы заболевания при ЮИА, необходимы два положительных результата теста с интервалом не менее 3 месяцев. 3
HLA-B27 обнаруживается у 14–42% пациентов с ЮИА в различных популяциях. 11,12 Точная роль HLA-B27 при ЮИА до конца не изучена. Его можно найти во всех категориях ЮИА, но чаще всего он встречается при артрите, ассоциированном с энтезитом. Это связано с клиническими проявлениями энтезита и сакроилеита, а также с более тяжелым исходом. 13
В настоящее время продолжаются исследования новых биомаркеров активности заболевания, таких как цитокины и MRP8 / 14, в дополнение к иммуногенетическим маркерам, но пока не создано ни одного теста в качестве стандартного клинического инструмента для помощи в диагностике и категоризации , или прогноз исхода при ЮИА. 14-16 Синовиальная жидкость показывает количество лейкоцитов в диапазоне от 600 до 100000 клеток / мм 3 (от 0,6 до 100 × 10 9 / л).
Визуализация
Традиционно рентгенография была методом выбора при ЮИА, но могла лишь косвенно описать воспаление синовиальной оболочки и мягких тканей. Рентгенография не может определить наличие и степень воспаления или предвестников разрушения костей для прогнозирования необратимого повреждения. Таким образом, новые методы, такие как ультрасонография и магнитно-резонансная томография (МРТ), приобрели все большее значение при диагностике, а также при оценке воспалительной активности и реакции на лечение.Еще неизвестно, окажутся ли эти новые методы лучше, чем рентгенография, в оценке повреждений и прогнозировании результатов. У детей с возрастом увеличивается окостенение хряща. 17 Основная проблема детской ревматологической визуализации — дифференцировать ранние признаки постоянного разрушения костей и хрящей от нормальных изменений в составе костного мозга, росте и созревании скелета.
Ранние рентгенологические изменения при ЮИА включают отек мягких тканей вокруг пораженных суставов, околосуставную остеопению и образование новой надкостницы (рис.101.1), что приводит к расширению средней части фаланг. В отличие от РА у взрослых, эрозии появляются поздно, потому что большая часть хряща в растущих суставах не кальцинирована, и визуализация эрозий возможна только после того, как хрящ кальцинирован. Сужение суставной щели — это ранний, но косвенный признак разрушения хряща. Эрозии или сужение суставной щели можно обнаружить уже через пару лет от начала активного артрита у ребенка с системным или полиартикулярным ЮИА.Даже при тяжелом ЮИА разрушенный хрящ можно заменить фиброхрящом, если болезнь стихнет (рис. 101.2). Линии остановки роста могут возникать в результате временной потери скорости роста и могут быть видны вокруг колена. Крестцово-подвздошные суставы у маленьких пациентов трудно интерпретировать из-за открытого эпифиза в крестцово-подвздошных суставах у растущих детей. Были предложены системы радиографической оценки, специфичные для ЮИА. 18,19
МРТ может быть отличным методом для демонстрации структуры суставного хряща, жидкости и мягких тканей в суставе.Последовательности с контрастным усилением ценны, потому что они могут продемонстрировать синовит (рис. 101.3). 20 Динамическая МРТ может использоваться для демонстрации сакроилеита у детей с клиническими характеристиками, соответствующими спондилоартриту. 20 В настоящее время для улучшения визуализации соответствующей ткани используется все больше методов МРТ с различной последовательностью импульсов, но остаются проблемы с определением и стандартизацией нормальных изменений развития у детей. 17 Пока нет надежной системы оценки синовита при ЮИА на основе МРТ, поскольку опубликованные на данный момент системы показали высокую вариабельность между наблюдателями. 21 МРТ также дорога и требует много времени, а у маленьких детей часто требуется общая анестезия.
Ультрасонография, которую можно проводить у постели больного, помогает визуализировать излияния в суставы, синовит и другие воспаления мягких тканей, включая энтезит, и имеет то преимущество, что ее легче выполнять без общей анестезии. 20 Его можно использовать для направления иглы для внутрисуставных инъекций, а с функцией энергетического допплера можно отслеживать синовиальное воспаление во время течения заболевания и вмешательств (рис.101,4). 22 Оценка воспаления и эрозии в развивающемся и созревающем скелете и мягких тканях остается особой проблемой для детей, и стандартизация и определения, используемые во взрослой ревматологии, не всегда подходят. 23
Границы | Воспаление синовиальной ткани, опосредованное аутоиммунными Т-клетками
Введение
Ревматоидный артрит (РА), которым страдает 1% населения во всем мире, представляет собой системное аутоиммунное заболевание, характеризующееся хроническим воспалением синовиальной ткани и разрушением суставов (1).Накапливается доказательство того, что различные типы иммунных клеток (например, Т-лимфоциты, В-клетки, макрофаги и нейтрофилы) с провоспалительным или противовоспалительным потенциалом задействуются в суставах после ревматоидного артрита и играют ключевую роль в развитии и прогрессе ревматоидного артрита (1). . Хотя молекулярные и клеточные механизмы патогенеза РА по-прежнему спорны, в настоящее время общепринято, что Т-хелперные (Th) клетки CD4 + играют решающую роль в проявлении болезни, о чем свидетельствует обильная инфильтрация Th-клеток в суставы, находящиеся при ревматоидном артрите. человеческий лейкоцитарный антиген (HLA) -DRB1, идентифицированный как ген с наибольшим риском развития РА, и высокая эффективность лечения CTLA4-Ig у пациентов с РА (1–3).С другой стороны, очевидно, что синовиоциты, подобные макрофагам и фибробластам (FLS), воспалительные цитокины, такие как TNFα, IL-1 и IL-6, присутствуют в синовиальной жидкости при RA и участвуют в воспалении и разрушении суставов. , что подтверждается высокой эффективностью антицитокиновой терапии, нацеленной на TNFα и IL-6, для контроля активности заболевания (4, 5). Однако то, как Th-клетки взаимодействуют с макрофагами и FLS или даже другими иммунными клетками, чтобы управлять хроническим синовиальным воспалением, недостаточно изучено.В этом обзоре мы обсуждаем, как аутоиммунные клетки Th27 побуждают FLS и врожденные лимфоидные клетки (ILC) в синовиальной ткани инициировать и поддерживать аутоиммунный артрит у мышей SKG (6), животной модели РА, как артритогенные клетки Th27 дифференцируются в сторону патогенных GM. -CSF-продуцирующие клетки при RA, и как регуляторные T (Treg) клетки становятся функционально нестабильными в воспаленных суставах RA.
Th-клеточно-зависимый аутоиммунный артрит у мышей SKG
Существует несколько моделей аутоиммунного артрита на мышах, которые внесли значительный вклад в наше понимание патогенетических механизмов РА.Например, мыши с коллаген-индуцированным артритом (CIA) и мыши K / BxN сильно зависят от выработки аутоантител против коллагена типа II и глюкозо-6-фосфат-изомеразы, соответственно, поскольку только сыворотка без клеточных компонентов от мышей с артритом является способны передавать болезнь мышам-реципиентам (7–9). Напротив, самореактивные клетки Th, но не аутоантитела, опосредуют воспаление суставов у мышей SKG, что делает мышей подходящей моделью для понимания того, как аутоиммунные Т-клетки инициируют и опосредуют синовиальное воспаление.
Штамм мышей SKG на фоне BALB / c спонтанно развивает опосредованный Th-клетками аутоиммунный артрит как следствие рецессивной точечной мутации в гене, кодирующем ζ-ассоциированный белок-70 (Zap-70), ключевую передачу сигналов, проксимальных к TCR. молекула (6, 10). Эта мутация нарушает положительный и отрицательный отбор Т-клеток в тимусе, приводя к продукции самореактивных Т-лимфоцитов CD4 + , включая артритогенные Th-клетки, и снижает продукцию Treg-клеток тимусом. Возникающий в результате аутоиммунный артрит напоминает РА человека в иммунопатологии, например, обильной инфильтрацией Th-клеток в артритные суставы и активным образованием аутоантител, таких как ревматоидный фактор и антитела к цитруллинированному пептиду (6, 10, 11).Спонтанный артрит у мышей SKG возникает в обычных условиях, но не в условиях отсутствия специфических патогенов (10, 12). Тем не менее, артрит может быть вызван активацией врожденного иммунитета через Toll-подобные рецепторы, путь дектина или пути комплемента, например, путем инъекции зимозана, неочищенного экстракта грибов, содержащего β-глюканы. Он также индуцируется путем обеспечения гомеостатической пролиферации артритогенных Т-лимфоцитов CD4 + у лимфопенических мышей, таких как мыши Rag2 — / — (12, 13).Недавно мы идентифицировали 60S рибосомный белок L23a (Rpl23a) в качестве артритогенного аутоантигена у мышей SKG с помощью анализа репертуара одноклеточных TCR эффекторных Th-клеток, выделенных из воспаленных суставов, а затем скрининга артритогенных потенциалов изолированных TCR у ретрогенных мышей, экспрессирующих один TCR. Более того, аутоантитела против RPL23A были специфически обнаружены в сыворотке от пациентов с РА, что дополнительно иллюстрирует сходство молекулярной основы воспаления суставов между артритом SKG и RA человека (14).Кроме того, сообщалось, что мыши SKG являются моделью болезни спондилоартрита, поскольку у них развиваются энтезит, дактилит, односторонний увеит, артрит крестцово-подвздошных суставов и воспаление позвонков вместе с артритом периферических суставов, которые являются клиническими признаками спондилоартрита (15, 16).
Среди субпопуляций Th-клеток мы показали, что продуцирующие IL-17 Т-хелперные (Th27) клетки играют ключевую роль в развитии артрита у мышей SKG (17). Перенос клеток CD4 + Т-клеток от мышей Il17a — / — SKG мышам с дефицитом Т-клеток полностью не индуцировал артрит. Il6 — / — У мышей SKG, у которых была нарушена дифференцировка Т-клеток в клетки Th27, практически не развился артрит. Хотя клетки Th2 и Th27 были обнаружены в воспаленных суставах мышей SKG, дефицит IFN-γ не улучшил, а скорее усугубил артрит SKG, поскольку дефицит IFN-γ усилил дифференцировку и патогенную функцию клеток Th27. Аналогичные результаты у мышей с дефицитом IFN-γ или его рецептором-дефицитом были также сделаны на других моделях Th27-опосредованного заболевания, таких как CIA и экспериментальный аутоиммунный энцефаломиелит (EAE), хорошо зарекомендовавшая себя модель рассеянного склероза на мышах (18, 19).Этот противовоспалительный эффект IFN-γ можно объяснить, по крайней мере частично, снижением экспрессии индоламин-2,3-диоксигеназы (IDO), катаболического фермента, ответственного за кинурениновый продукт триптофана. IDO экспрессируется в различных типах клеток, включая макрофаги и дендритные клетки (DC), и индуцируется во время воспаления IFN-γ или другими воспалительными цитокинами. Повышенные уровни IFN-γ-опосредованной экспрессии IDO в ДК в воспаленных тканях играют ключевую роль в иммунной регуляции, отчасти за счет лишения местного триптофана с последующей активацией общего контрольного нерепрессированного пути 2 киназы в ответ на Т-клетки, что приводит к к подавлению активации и пролиферации местных Т-клеток, включая патогенные клетки Th27.Напротив, дефицит IFN-γ подавляет экспрессию IDO, что впоследствии позволяет патогенным клеткам Th27 расширяться в местах воспаления и усиливать аутоиммунные патологии (20–23). Эти данные, взятые вместе, подтверждают, что мыши SKG являются подходящей животной моделью для изучения того, как аутоиммунные Т-клетки организуют хроническое воспаление суставов (17).
Инициирование артрита взаимодействием между артритогенными клетками Th27 и фибробластоподобными синовиоцитами
Для инициации артрита у мышей SKG требуются аутореактивные (артритогенные) CD4 + Т-клетки, чтобы дифференцироваться в эффекторные клетки Th27 и мигрировать в суставы.В качестве механизма этой дифференцировки Th27 аутореактивных CD4 + Т-клеток они сначала активируются на периферии за счет сильного распознавания комплексов MHC класса II / ауто-пептид, экспрессируемых антигенпрезентирующими клетками (APC), и реципрокно стимулируют APC повышают регуляцию CD80 / CD86 посредством взаимодействия CD40 / CD40L, дополнительно усиливая активацию и пролиферацию самореактивных Т-клеток CD4 + . Таким образом, APC стимулировали секрецию IL-6, IL-1 и IL-23, которые вместе с местным тканевым TGFβ управляют дифференцировкой наивных CD4 + Т-клеток в артритогенные клетки Th27.Миграция артритогенных клеток Th27 в синовиальную ткань происходит хемокин-зависимым образом, в частности, через ось CCR6-CCL20. То есть клетки Th27 преимущественно экспрессируют CCR6, тогда как активированные FLS секретируют CCL20, лиганд CCR6, так что FLS предпочтительно рекрутируют клетки Th27 в синовиальную ткань. Фактически, моноклональные антитела, блокирующие CCR6, значительно снижали тяжесть артрита у мышей SKG за счет ингибирования инфильтрации клеток Th27 в суставы (24). Более того, FLS усиливают экспрессию CCL20 в ответ на IL-17, IL-1 или TNFα, тогда как IFN-γ или IL-4 ингибируют экспрессию (24).Эти наблюдения иллюстрируют важное прямое взаимодействие между клетками Th27 и FLS в инициировании и усилении воспаления суставов: как только артритогенные клетки Th27 активируются и рекрутируются в суставы для инициирования воспаления, FLS дополнительно увеличивают CCL20 в ответ на IL-17 и другие провоспалительные цитокины и ускоряют набор клеток Th27 для инициирования и усиления артрита (24). Активированные FLS взаимодействуют не только с клетками Th27, но и с другими иммунными клетками, вызывая хроническое синовиальное воспаление, например, путем усиления секреции хемокинов, таких как Cxcl1 и Cxcl5, для набора нейтрофилов в суставы (25).Интересно, что аналогично этим находкам у мышей SKG, клетки Th27 и синовиоциты у пациентов с РА экспрессируют CCR6 и CCL20, соответственно, со значительной корреляцией между уровнем IL-17 и CCL20 в суставах с RA (24). Кроме того, недавние исследования GWAS идентифицировали ген CCR6 как локус гена восприимчивости к заболеванию RA. Эти наблюдения в совокупности указывают на взаимодействие между клетками Th27 и FLS как ключевым механизмом развития воспаления синовиальной ткани (26).
GM-CSF, ключевой медиатор, поддерживающий хроническое воспаление суставов
Гранулоцитарный макрофагальный колониестимулирующий фактор (GM-CSF) из различных источников в воспаленных синовиальных тканях усиливает хроническое синовиальное воспаление.Недавно мы продемонстрировали, что GM-CSF из лимфоидных и тканевых резидентных клеток сустава является ключевым компонентом для инициации и поддержания аутоиммунного артрита (25). GM-CSF недавно был выделен как мощный провоспалительный цитокин, который активирует моноциты и макрофаги и вырабатывается различными типами клеток, например фибробластами, эндотелиальными клетками и Т-клетками, в ответ на воспалительные стимулы (27). GM-CSF может быть нацелен на лечение RA, поскольку он в изобилии в синовиальной оболочке RA в сочетании с сильной активацией синовиальных макрофагов и активной выработкой IL-6 и TNFα в суставах RA (28, 29).Кроме того, у мышей Csf2 — / — SKG полностью не развился артрит независимо от присутствия эффекторных клеток Th27 на периферии, что указывает на незаменимую роль GM-CSF в артрите SKG (25). GM-CSF частично продуцировался эффекторными клетками Th27 в воспаленных суставах; однако GM-CSF, полученный из Т-клеток, хотя и усиливает артрит, был незаменим для индукции артрита, в то время как GM-CSF, полученный не из Т-клеток, был незаменим. Создавая смешанные химеры костного мозга, мы определили радиорезистентные стромальные клетки, включая FLS и синовиальные резидентные ILC, в качестве важнейшего источника GM-CSF.Действительно, тяжесть артрита в химерах костного мозга была значительно снижена, когда продукция GM-CSF была специфически ингибирована или потеряна либо в радиоустойчивых стромальных клетках, либо в ILC. Повторная стимуляция IL-17 индуцировала продукцию GM-CSF из FLS in vitro . Более того, когда Th-клетки от мышей SKG дикого типа, но не мышей Il17a — / — SKG, были адоптивно перенесены в мышей Rag2 — / — , синовиоциты значительно увеличили экспрессию Csf2 вместе с Il6 и Ccl20 .Эти наблюдения, взятые вместе, предполагают, что IL-17 из эффекторных клеток Th27, мигрирующих в синовиальную ткань, стимулирует FLS для производства GM-CSF. Небольшое количество ILC присутствует в здоровых суставах мышей SKG, а также в других линиях здоровых мышей; среди них ИЛК, продуцирующие GM-CSF, значительно расширились в воспаленных суставах. Интересно, что большая часть этих синовиальных ILC, продуцирующих GM-CSF, обладала характеристиками ILC группы 2 (ILC2). Например, они преимущественно экспрессируют GATA3 и / или IL-13, известные как главный фактор транскрипции и сигнатурный цитокин, соответственно, ILC2s, и повышают продукцию in vitro GM-CSF, а также IL-5 и IL- 13 в ответ на комбинацию стимуляции IL-2 и IL-33.Кроме того, эти синовиальные ILC сильно экспрессируют Toll-подобный рецептор 9 (Tlr9) и синергетически увеличивают продукцию GM-CSF при стимуляции комбинацией IL-33 и ДНК CpG, лиганда TLR9, но не только ДНК CpG. Таким образом, вероятно, что синовиальные ILC могут отвечать на сигналы от некротических клеток в воспаленных суставах, такие как алармины и молекулярные паттерны, связанные с повреждениями (DAMP), включая IL-33 и митохондриальную ДНК. Эти данные, вместе взятые, показывают, что, в отличие от FLS, синовиальные ILC активируются, увеличивают и увеличивают производство GM-CSF, воспринимая сигналы окружающей среды, такие как IL-2, IL-33 и собственная ДНК, которые могут быть получены из артритогенных клеток Th27 и поврежденные клетки в воспаленных суставах (25).
Таким образом, при аутоиммунном артрите у мышей SKG, который полностью зависит от клеток Th27, ключевым первичным событием в инициировании артрита является миграция клеток Th27 в синовиальную ткань, чтобы стимулировать FLS с помощью IL-17 и модулировать их воспалительные профили. . После инициации артрита артритогенные клетки Th27 вместе с FLS и синовиальными ILC организуют воспалительный каскад для усиления хронического воспаления суставов с помощью GM-CSF из различных источников, подверженных различным иммунологическим стимулам, что приводит к активации синовиальных макрофагов и их обильной продукции pro — воспалительные цитокины, такие как IL-1, IL-6 и TNFα (рис. 1).
Рисунок 1 . Хроническое воспаление суставов, опосредованное FLS и воспалительными иммунными клетками при артрите SKG. Артритогенные клетки Th27 мигрируют в синовиальную ткань и инициируют воспаление суставов, стимулируя FLS через IL-17. Активированные FLS секретируют различные медиаторы воспаления, чтобы задействовать и расширить различные воспалительные иммунные клетки. На хронической стадии клетки FLS, ILC2 и Th27 продуцируют GM-CSF для активации воспалительных моноцитов и облегчения хронического воспаления суставов.
Th-клетки при ревматоидном артрите
До открытия клеток Th27, RA считался Th2-опосредованным аутоиммунным заболеванием, основанным на классической парадигме Th2 / Th3 в аутоиммунитете. Действительно, IFN-γ + Th-клетки широко распространены среди CD4 + Т-клеток в синовиальной жидкости (SF) при RA (30, 31). Однако патогенная роль IFN-γ при РА была неясной, поскольку вмешательство IFN-γ в клинических испытаниях дало противоречивые результаты (32–37). После открытия клеток Th27 и их решающей роли в моделях на животных различных аутоиммунных заболеваний, включая аутоиммунный артрит, роль IL-17 и подмножества Th27 при РА была тщательно изучена.Однако SF RA содержит только низкие уровни IL-17 и лишь небольшое количество клеток Th27 (38–41). Более того, клинические испытания фазы III с использованием ингибиторов IL-17 у пациентов с РА не показали значительного преимущества по сравнению с одобренными в настоящее время биологическими агентами (42, 43). Эти клинические результаты предполагают, что одного вмешательства IL-17 может быть недостаточно для облегчения хронического воспаления суставов при РА. Недавние исследования пластичности клеток Th27 при РА и ювенильном идиопатическом артрите (ЮИА) могут дать возможное объяснение этим наблюдениям.То есть сообщалось, что значительная популяция Т-клеток IFN-γ + в SF коэкспрессирует CCR6 и CD161, поверхностные маркеры клеток Th27 человека, и, следовательно, клетки Th27 могут быть преобразованы в Th2-подобный фенотип. через промежуточное состояние IFN-γ + IL-17 + Т-клетки, когда они сталкиваются со средой IL-12 high , что наблюдается в SF при RA и JIA (44, 45). Такие фенотипические изменения клеток Th27 в суставах RA могут объяснить неудачу клинических испытаний с использованием ингибиторов IL-17.
Ex-Th27 клеток в суставах RA, по-видимому, критически способствуют прогрессу RA, по крайней мере, частично за счет продукции GM-CSF (46). Патогенная роль GM-CSF при РА была изучена, и недавние клинические испытания с использованием ингибиторов GM-CSF показали значительную эффективность (47–50). Таким образом, вероятно, что патогенные клетки Th27 могут играть разные роли на разных фазах РА: продуцирующие IL-17 клетки Th27 для инициации на ранней фазе и клетки ex-Th27, продуцирующие GM-CSF, для хронического воспаления на более поздней фазе.После инициации артрита большинство клеток Th27 превращаются в клетки IFN-γ + ex-Th27 в ответ на среду, богатую IL-12 в суставах, пораженных артритом, и начинают активно продуцировать GM-CSF вместе с другими GM-CSF- продуцирующие клетки, такие как FLS и синовиальные ILC. Обогащенная GM-CSF среда в SF приводит к активации синовиальных макрофагов и индукции большого количества провоспалительных цитокинов, таких как IL-1, IL-6 и TNFα, что приводит к хроническому разрушению кости. Пластичность клеток Th27 по отношению к Th2-подобному фенотипу редко наблюдается на модели артрита SKG, но ее можно увидеть на других моделях аутоиммунного заболевания на мышах (25).Например, картирование судеб IL-17 у мышей показало, что высокопатогенные Т-клетки IFN-γ + или GM-CSF + в спинном мозге мышей EAE происходят исключительно из клеток Th27 (51). Есть надежда, что дальнейшее выяснение молекулярных механизмов пластичности Th27 на аутоиммунных моделях поможет разработать эффективные способы контроля патогенных ex-Th27 клеток при RA.
Эффект передачи сигналов TNFα на патогенные Т-клетки считается стимулирующим, но кинетика Th27-клеток у пациентов с РА после лечения ингибиторами TNF, по-видимому, непостоянна (52).Отношение Th27-клеток к регуляторным T (Treg) -клеткам у пациентов с РА было значительно выше, чем у здоровых контролей (53). Стимуляция in vitro TNFα прямо и косвенно способствовала дифференцировке Th27, что согласуется с корреляцией терапевтической эффективности ингибиторов TNF у пациентов с РА со сниженным соотношением Th27 / Treg в периферической крови (54-57). Однако у пациентов с РА, которые не ответили на лечение ингибиторами TNF, частота Th27-клеток в их периферической крови парадоксальным образом увеличилась после лечения (57, 58).Механизмы, лежащие в основе этого парадоксального клинического исхода, остаются неясными, но это может быть связано с различными иммунологическими эффектами передачи сигналов TNFα через рецептор TNF 1 (TNFR1) и TNFR2, а также воспалительные клетки, экспрессирующие разные уровни этих рецепторов (52). Например, специфическое устранение передачи сигналов TNFα через TNFR1, но не через TNFR2, усиливало экспрессию IL-12 / IL-23 p40 в макрофагах, управляя ответами Th27 (59). Точно так же моноциты пациентов с РА, не отвечающих на лечение, которые показали повышенный ответ Th27 после лечения ингибиторами TNF, продуцировали больше IL-12 / IL-23 p40 (58).Эти данные, вместе взятые, предполагают, что терапевтические эффекты ингибиторов TNF на хроническое воспаление суставов у пациентов с РА различны из-за их гетерогенных клеточных ответов через передачу сигналов TNFR1 и TNFR2 и вариабельного состава синовиоцитов и их экспрессии двух рецепторов.
Регуляторные Т-клетки в РА
КлеткиTreg, экспрессирующие фактор транскрипции Foxp3, незаменимы для поддержания иммунологической толерантности и подавления развития различных аутоиммунных заболеваний, включая аутоиммунный артрит (60).Однако имеется мало сообщений о взаимодействиях между Treg-клетками и FLS как на моделях RA человека, так и на животных. Treg-клетки при аутоиммунном артрите обсуждались в связи с нестабильностью и нарушением функции Treg-клеток при воспалительных условиях в воспаленных суставах (61). Среди провоспалительных цитокинов IL-6 является хорошо охарактеризованным цитокином, который определяет баланс дифференцировки Th27 / Treg-клеток на периферии (54, 62–64). Действительно, лечение активного RA с помощью тоцилизумаба, гуманизированного антитела против рецептора IL-6, значительно снижает клиническую активность заболевания наряду со значительным уменьшением количества Th27-клеток и увеличением частоты Treg-клеток в периферической крови (65).Это указывает на то, что высокие уровни системного IL-6 у пациентов с РА склоняют чашу весов в сторону доминирования клеток Th27. Обильный IL-6 в SF RA, в основном продуцируемый FLS и синовиальными макрофагами, также может влиять на дифференцировку и разрастание случайных Th-клеток, инфильтрирующих артритные суставы.
Иммуномодулирующая роль TNFα в стабильности и подавляющих функциях Treg-клеток остается спорной. Первоначально сообщалось о дефектной функции Treg-клеток в среде, богатой TNFα, что наблюдается в периферической крови и артритных суставах при РА, и было предложено несколько возможных механизмов.Например, высокие уровни TNFα могут предотвращать функцию Treg, ингибируя образование иммунологического синапса (IS) между Treg и APC. Динамика протеинкиназы Cθ (PKC-θ) в IS, по-видимому, реципрокно регулируется в эффекторных T- и Treg-клетках: рекрутирование PKC-θ в IS было необходимо для полной активации эффекторных T-клеток, тогда как рекрутирование PKC-θ уменьшалось. подавляющая активность Treg-клеток. Примечательно, что высокие уровни TNFα усиливают рекрутирование PKC-θ в IS в Treg-клетках и ингибируют их супрессивную активность, тогда как блокада PKC-θ защищает Treg-клетки от инактивации TNFα и восстанавливает супрессивную функцию (66).Напротив, большой дисковый гомолог 1 (Dlgh2), каркасный белок, рекрутировался в IS в 4 раза больше в Treg-клетках, чем в эффекторных Т-клетках, что усиливало подавляющую функцию Treg-клеток. Клетки Treg от активных пациентов с РА или от здоровых людей из контрольной группы, получавших TNFα, показали снижение рекрутирования Dlgh2 и снижение подавляющей функции. Кроме того, подавление экспрессии гена Dlgh2 нарушает функцию Treg-клеток человека (67). Кроме того, подавляющая функция Treg-клеток в присутствии TNFα модулируется дефосфорилированием в сайте Ser418 FOXP3 через протеинфосфатазу 1, и, напротив, лечение антителами против TNFα восстанавливает функцию Treg у пациентов с РА (68).Эти данные в совокупности указывают на то, что хроническое воспаление ингибирует супрессивную функцию Treg-клеток при RA отчасти из-за высоких уровней TNFα, которые модулируют фосфорилирование FOXP3, а также регулируют экспрессию и перемещение PKC-θ и Dlgh2 присущим Treg клеткам. Однако недавно эта концепция была оспорена. Согласно сообщениям с различными экспериментальными условиями, TNFα не подавлял подавляющую функцию человеческих Treg-клеток, а скорее усиливал экспрессию CD25 и Foxp3 в присутствии IL-2, а также способствовал пролиферации и выживанию Treg-клеток посредством прямого влияние TNFα на TNFR 2 присущим Treg клеткам, чтобы контролировать эффекторные Т-клетки (69, 70).Влияние TNFα на эффекторные Т-клетки также следует учитывать, как упомянуто выше, и эффекторные T-субпопуляции в среде с высоким содержанием TNF могут быть более устойчивыми к подавлению Treg-клетками, по крайней мере частично, из-за костимулирующих эффектов TNFα на эффекторные Т-клетки. Для устранения этого несоответствия необходимы дальнейшие исследования.
Заключительное замечание
Биологические агенты изменили парадигму лечения РА. Однако до сих пор остается неясным, как антицитокиновые агенты модулируют воспалительные свойства Т-клеток, FLS и ILC в синовиальной ткани в ходе лечения, и как лечение влияет на взаимодействия между эффекторными иммунными клетками и FLS при хроническом воспалении суставов.Модели на животных, такие как мыши SKG, полезны для решения этих проблем. Недавние исследования, включая наше собственное, действительно выявили роль аутоантител против RPL23A и GM-CSF, обычно обнаруживаемых у мышей SKG и пациентов с РА. Исследования также выявили ранее недооцененный аспект воспалительного каскада, опосредованного артритогенными клетками Th27, взаимодействующими с кроветворными и негематопоэтическими клетками синовиальной ткани. Дальнейшие исследования помогут нам понять молекулярную и клеточную основу хронического воспаления суставов и помогут разработать новое лечение РА с большей специфичностью и эффективностью.
Авторские взносы
YT, KH и SS написали и отредактировали рукопись.
Заявление о конфликте интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Благодарности
Эта работа была поддержана JSPS Grants-in Aid for Scientific Research (16H06233 для KH и 16H06295 для SS).
Список литературы
2.Грегерсен П.К., Сильвер Дж., Винчестер Р.Дж. Гипотеза общего эпитопа. Подход к пониманию молекулярной генетики предрасположенности к ревматоидному артриту. Arthritis Rheum. (1987) 30: 1205–13. DOI: 10.1002 / art.1780301102
PubMed Аннотация | CrossRef Полный текст | Google Scholar
3. Gonzalez-Gay MA, Garcia-Porrua C, Hajeer AH. Влияние лейкоцитарного антигена DRB1 человека на восприимчивость и тяжесть ревматоидного артрита. Semin Arthritis Rheum. (2002) 31: 355–60. DOI: 10.1053 / sarh.2002.32552
PubMed Аннотация | CrossRef Полный текст | Google Scholar
6. Сакагучи Н., Такахаши Т., Хата Х., Номура Т., Тагами Т., Ямадзаки С. и др. Измененный отбор Т-клеток тимуса из-за мутации гена ZAP-70 вызывает аутоиммунный артрит у мышей. Природа. (2003) 426: 454–60. DOI: 10.1038 / nature02119
PubMed Аннотация | CrossRef Полный текст | Google Scholar
7. Куртенэ Дж. С., Даллман М. Дж., Даян А. Д., Мартин А., Мозедейл Б.Иммунизация против гетерологичного коллагена типа II вызывает артрит у мышей. Природа. (1980) 283: 666–8. DOI: 10.1038 / 283666a0
PubMed Аннотация | CrossRef Полный текст | Google Scholar
8. Мацумото И., Стауб А., Бенуа С., Матис Д. Артрит, спровоцированный сцепленным распознаванием гликолитического фермента Т- и В-клетками. Наука. (1999) 286: 1732–5. DOI: 10.1126 / science.286.5445.1732
PubMed Аннотация | CrossRef Полный текст | Google Scholar
9.Мацумото И., Макчиони М., Ли Д.М., Морис М., Симмонс Б., Бреннер М. и др. Как антитела к вездесущему цитоплазматическому ферменту могут спровоцировать специфическое для суставов аутоиммунное заболевание. Nat Immunol. (2002) 3: 360–5. DOI: 10.1038 / ni772
PubMed Аннотация | CrossRef Полный текст | Google Scholar
10. Хата Х, Сакагути Н., Ёситоми Х, Ивакура Й, Секикава К., Адзума Й и др. Отчетливый вклад ИЛ-6, ФНО-альфа, ИЛ-1 и ИЛ-10 в Т-клеточный спонтанный аутоиммунный артрит у мышей. J Clin Invest. (2004) 114: 582–8. DOI: 10.1172 / JCI21795
PubMed Аннотация | CrossRef Полный текст | Google Scholar
11. Танака С., Маеда С., Хашимото М., Фудзимори С., Ито Й, Терадаира С. и др. Постепенное ослабление передачи сигналов TCR вызывает различные аутоиммунные заболевания за счет изменения отбора Т-клеток тимуса и регуляторной функции Т-клеток. J Immunol. (2010) 185: 2295–305. DOI: 10.4049 / jimmunol.1000848
PubMed Аннотация | CrossRef Полный текст | Google Scholar
12.Ёситоми Х., Сакагучи Н., Кобаяси К., Браун Г.Д., Тагами Т., Сакихама Т. и др. Роль грибковых {бета} -глюканов и их рецептора Dectin-1 в индукции аутоиммунного артрита у генетически восприимчивых мышей. J Exp Med. (2005) 201: 949–60. DOI: 10.1084 / jem.20041758
PubMed Аннотация | CrossRef Полный текст | Google Scholar
13. Хашимото М., Хирота К., Йошитоми Х., Маеда С., Терадаира С., Акизуки С. и др. Комплемент управляет дифференцировкой клеток Th27 и запускает аутоиммунный артрит. J Exp Med. (2010) 207: 1135–43. DOI: 10.1084 / jem.20092301
PubMed Аннотация | CrossRef Полный текст | Google Scholar
14. Ито Й, Хашимото М., Хирота К., Окура Н., Морикава Х., Нисикава Х. и др. Обнаружение Т-клеточного ответа на повсеместно распространенный клеточный белок при аутоиммунном заболевании. Наука. (2014) 346: 363–8. DOI: 10.1126 / science.1259077
PubMed Аннотация | CrossRef Полный текст | Google Scholar
16. Ruutu M, Thomas G, Steck R, Degli-Esposti MA, Zinkernagel MS, Alexander K, et al.Бета-глюкан вызывает спондилоартрит и илеит, подобный болезни Крона, у мышей SKG. Arthritis Rheum. (2012) 64: 2211–22. DOI: 10.1002 / art.34423
PubMed Аннотация | CrossRef Полный текст | Google Scholar
17. Хирота К., Хашимото М., Ёситоми Х., Танака С., Номура Т., Ямагути Т. и др. Самореактивность Т-клеток формирует цитокиновую среду для спонтанного развития IL-17 + Th-клеток, которые вызывают аутоиммунный артрит. J Exp Med. (2007) 204: 41–7. DOI: 10.1084 / jem.20062259
PubMed Аннотация | CrossRef Полный текст | Google Scholar
18. Манури-Шварц Б., Чиоккиа Г., Бессис Н., Абехсира-Амар О., Батте Ф., Мюллер С. и др. Высокая восприимчивость к коллаген-индуцированному артриту у мышей, лишенных рецепторов IFN-гамма. J Immunol. (1997) 158: 5501–6.
PubMed Аннотация | Google Scholar
19. Chu CQ, Wittmer S, Dalton DK. Неспособность подавить рост популяции активированных Т-лимфоцитов CD4 у мышей с гамма-дефицитом интерферона приводит к обострению экспериментального аутоиммунного энцефаломиелита. J Exp Med. (2000) 192: 123–8. DOI: 10.1084 / jem.192.1.123
PubMed Аннотация | CrossRef Полный текст | Google Scholar
20. Курти А., Трабанелли С., Сальвестрини В., Баккарани М., Лемоли Р.М. Роль индоламин-2,3-диоксигеназы в индукции иммунной толерантности: акцент на гематологии. Кровь. (2009) 113: 2394–401. DOI: 10.1182 / кровь-2008-07-144485
PubMed Аннотация | CrossRef Полный текст | Google Scholar
21. Манн Д.Х., Шарма М.Д., Бабан Б., Хардинг Х.П., Чжан Й., Рон Д. и др.Киназа GCN2 в Т-клетках опосредует остановку пролиферации и индукцию анергии в ответ на индоламин-2,3-диоксигеназу. Иммунитет. (2005) 22: 633–42. DOI: 10.1016 / j.immuni.2005.03.013
PubMed Аннотация | CrossRef Полный текст | Google Scholar
22. Criado G, Simelyte E, Inglis JJ, Essex D, Williams RO. Катаболизм триптофана, опосредованный индолеамин-2,3-диоксигеназой, регулирует накопление клеток Th2 / Th27 в суставе при артрите, индуцированном коллагеном. Arthritis Rheum. (2009) 60: 1342–51. DOI: 10.1002 / art.24446
PubMed Аннотация | CrossRef Полный текст | Google Scholar
23. Ли Дж., Ли Дж., Парк М.К., Лим М.А., Парк Е.М., Ким Е.К. и др. Гамма-интерферон подавляет коллаген-индуцированный артрит за счет регуляции Th27 за счет индукции индоламин-2,3-дезоксигеназы. PLoS ONE. (2013) 8: e60900. DOI: 10.1371 / journal.pone.0060900
PubMed Аннотация | CrossRef Полный текст | Google Scholar
24. Хирота К., Ёситоми Х., Хашимото М., Маеда С., Терадаира С., Сугимото Н. и др.Предпочтительное привлечение CCR6-экспрессирующих клеток Th27 к воспаленным суставам через CCL20 при ревматоидном артрите и его животной модели. J Exp Med. (2007) 204: 2803–12. DOI: 10.1084 / jem.20071397
PubMed Аннотация | CrossRef Полный текст | Google Scholar
25. Хирота К., Хашимото М., Ито Й., Мацуура М., Ито Х., Танака М. и др. Аутоиммунные клетки Th27 индуцировали секрецию цитокина GM-CSF синовиальными стромальными и врожденными лимфоидными клетками, чтобы инициировать и усиливать аутоиммунный артрит. Иммунитет. (2018) 48: 1220–32 e5. DOI: 10.1016 / j.immuni.2018.04.009
PubMed Аннотация | CrossRef Полный текст | Google Scholar
26. Кочи Ю., Окада Ю., Сузуки А., Икари К., Терао С., Такахаши А. и др. Вариант регуляции CCR6 связан с восприимчивостью к ревматоидному артриту. Nat Genet. (2010) 42: 515–9. DOI: 10,1038 / нг.583
PubMed Аннотация | CrossRef Полный текст | Google Scholar
27. Gasson JC. Молекулярная физиология гранулоцитарно-макрофагального колониестимулирующего фактора. Кровь. (1991) 77: 1131–45.
PubMed Аннотация | Google Scholar
28. Альваро-Грасиа Дж. М., Звайфлер Н. Дж., Браун С. Б., Каушанский К., Файрштейн Г. С.. Цитокины при хроническом воспалительном артрите. VI. анализ синовиальных клеток, участвующих в производстве гранулоцитарно-макрофагального колониестимулирующего фактора и экспрессии генов при ревматоидном артрите и его регуляции с помощью IL-1 и фактора некроза опухоли-альфа. J Immunol. (1991) 146: 3365–71.
PubMed Аннотация | Google Scholar
29.Райт Х.Л., Бакнелл Р.К., Мутс Р.Дж., Эдвардс SW. Анализ цитокинов SF и плазмы дает представление о механизмах воспалительного артрита и может предсказать ответ на терапию. Ревматология. (2012) 51: 451–9. DOI: 10.1093 / ревматология / ker338
PubMed Аннотация | CrossRef Полный текст | Google Scholar
30. Ито Й, Усуи Т., Кобаяши С., Игучи-Хашимото М., Ито Х., Йошитоми Х. и др. Гамма / дельта Т-клетки являются основным источником интерлейкина-17 в пораженных суставах при коллаген-индуцированном артрите, но не при ревматоидном артрите. Arthritis Rheum. (2009) 60: 2294–303. DOI: 10.1002 / art.24687
CrossRef Полный текст | Google Scholar
31. Ямада Х., Накашима Ю., Окадзаки К., Маватари Т., Фукуши Дж. И., Кайбара Н. и др. В суставах пациентов с ревматоидным артритом преобладают клетки Th2, но не Th27. Ann Rheum Dis. (2008) 67: 1299–304. DOI: 10.1136 / ard.2007.080341
CrossRef Полный текст | Google Scholar
32. Firestein GS, Zvaifler NJ. Активация моноцитов периферической крови и синовиальной жидкости при воспалительном артрите.II. низкие уровни синовиальной жидкости и интерферона синовиальной ткани позволяют предположить, что гамма-интерферон не является основным фактором активации макрофагов. Arthritis Rheum. (1987) 30: 864–71. DOI: 10.1002 / art.1780300804
CrossRef Полный текст | Google Scholar
34. Сигидин Ю.А., Лукина Г.В., Скуркович Б., Скуркович С. Рандомизированное двойное слепое исследование антител к интерферон-гамма при ревматоидном артрите. Scand J Rheumatol. (2001) 30: 203–7. DOI: 10.1080 / 030097401316
0
PubMed Аннотация | CrossRef Полный текст | Google Scholar
35.Lemmel EM, Brackertz D, Franke M, Gaus W., Hartl PW, Machalke K и др. Результаты многоцентрового плацебо-контролируемого двойного слепого рандомизированного клинического исследования III фазы по лечению ревматоидного артрита рекомбинантным гамма-интерфероном. Rheumatol Int. (1988) 8: 87–93. DOI: 10.1007 / BF00271840
PubMed Аннотация | CrossRef Полный текст | Google Scholar
36. Cannon GW, Emkey RD, Denes A, Cohen SA, Saway PA, Wolfe F и др. Проспективное 5-летнее наблюдение за рекомбинантным гамма-интерфероном при ревматоидном артрите. J Rheumatol. (1993) 20: 1867–73.
Google Scholar
37. Стеттер С., Ауэр И.О., Папст С., Боескен У.Х., Штирль II Х.Э., Ботценхардт У. и др. Двойное слепое контролируемое многоцентровое клиническое исследование фазы III с применением гамма-интерферона при ревматоидном артрите. Немецкая группа по изучению лимфокинов. Rheumatol Int. (1992) 12: 175–85.
Google Scholar
38. Лейпе Дж., Грюнке М., Дечан С., Рейндл С., Керцендорф Ю., Шульце-Купс Х. и др. Роль клеток Th27 в аутоиммунном артрите человека. Arthritis Rheum. (2010) 62: 2876–85. DOI: 10.1002 / art.27622
PubMed Аннотация | CrossRef Полный текст | Google Scholar
39. Пенатти А., Фаччиотти Ф, Де Маттеис Р., Ларги П., Парони М., Мурго А. и др. Различия в профилях сывороточных и синовиальных CD4 + Т-клеток и цитокинов для стратификации пациентов с воспалительным остеоартритом и ревматоидным артритом. Arthritis Res Ther. (2017) 19: 103. DOI: 10.1186 / s13075-017-1305-1
PubMed Аннотация | CrossRef Полный текст | Google Scholar
40.van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, et al. Клетки Th27, но не клетки Th2, от пациентов с ранним ревматоидным артритом являются мощными индукторами матриксных металлопротеиназ и провоспалительных цитокинов при взаимодействии синовиальных фибробластов, включая выработку аутокринного интерлейкина-17A. Arthritis Rheum. (2011) 63: 73–83. DOI: 10.1002 / art.30093
CrossRef Полный текст | Google Scholar
41. Джандус К., Биолей Дж., Соперники Дж. П., Дудлер Дж., Шпайзер Д., Ромеро П.Повышенное количество циркулирующих полифункциональных клеток памяти Th27 у пациентов с серонегативными спондилоартритами. Arthritis Rheum. (2008) 58: 2307–17. DOI: 10.1002 / art.23655
PubMed Аннотация | CrossRef Полный текст | Google Scholar
42. Бланко Ф.Дж., Морике Р., Докупилова Е., Коддинг С., Нил Дж., Андерссон М. и др. Секукинумаб при активном ревматоидном артрите: рандомизированное, двойное слепое, активное исследование с использованием препаратов сравнения и плацебо III фазы. Arthritis Rheumatol. (2017) 69: 1144–53. DOI: 10.1002 / art.40070
PubMed Аннотация | CrossRef Полный текст | Google Scholar
43. Докупилова Е., Элион Дж., Такеучи Т., Малаволта Н., Сфикакис П.П., Ван Й. и др. Секукинумаб после терапии противоопухолевым фактором некроза-альфа: исследование III фазы активного ревматоидного артрита. Scand J Rheumatol. (2018) 47: 276–81. DOI: 10.1080 / 03009742.2017.13
PubMed Аннотация | CrossRef Полный текст | Google Scholar
44.Нистала К., Адамс С., Камбрук Х., Урсу С., Оливито Б., де Ягер В. и др. Пластичность Th27 при аутоиммунном артрите человека определяется воспалительной средой. Proc Natl Acad Sci USA. (2010) 107: 14751–6. DOI: 10.1073 / pnas.1003852107
PubMed Аннотация | CrossRef Полный текст | Google Scholar
45. Басдео С.А., Клакстон Д., Сулеймани Дж., Моран Б., Канаван М., Орр С. и др. Клетки Ex-Th27 (неклассические Th2) функционально отличаются от классических клеток Th2 и Th27 и не ограничиваются регуляторными Т-клетками. J Immunol. (2017) 198: 2249–59. DOI: 10.4049 / jimmunol.1600737
CrossRef Полный текст | Google Scholar
46. Пайпер С., Пезенакер А.М., Бендинг Д., Тиругнанабалан Б., Варсани Х., Веддерберн Л.Р. и др. Экспрессия Т-клетками колониестимулирующего фактора гранулоцитов-макрофагов при ювенильном артрите зависит от пластичности Th27. Arthritis Rheumatol. (2014) 66: 1955–60. DOI: 10.1002 / art.38647
PubMed Аннотация | CrossRef Полный текст | Google Scholar
47.Беренс Ф., Так П.П., Остергаард М., Стойлов Р., Виланд П., Хейзинга Т.В. и др. MOR103, человеческое моноклональное антитело к гранулоцитарно-макрофагальному колониестимулирующему фактору, в лечении пациентов с умеренным ревматоидным артритом: результаты фазы Ib / IIa рандомизированного, двойного слепого, плацебо-контролируемого исследования с увеличением дозы. Ann Rheum Dis. (2015) 74: 1058–64. DOI: 10.1136 / annrheumdis-2013-204816
PubMed Аннотация | CrossRef Полный текст | Google Scholar
48. Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, et al.Эффективность и безопасность маврилимумаба у пациентов с ревматоидным артритом. Ann Rheum Dis. (2013) 72: 1445–52. DOI: 10.1136 / annrheumdis-2012-202450
PubMed Аннотация | CrossRef Полный текст | Google Scholar
49. Бурместер Г.Р., Макиннес И.Б., Кремер Дж., Миранда П., Коркош М., Венковски Дж. И др. Рандомизированное исследование фазы IIb маврилимумаба, нового моноклонального антитела к рецептору GM-CSF альфа, в лечении ревматоидного артрита. Ann Rheum Dis. (2017) 76: 1020–30.DOI: 10.1136 / annrheumdis-2016-210624
PubMed Аннотация | CrossRef Полный текст | Google Scholar
50. Weinblatt ME, McInnes IB, Kremer JM, Miranda P, Vencovsky J, Guo X, et al. Рандомизированное исследование фазы IIb маврилимумаба и голимумаба при ревматоидном артрите. Arthritis Rheumatol. (2018) 70: 49–59. DOI: 10.1002 / art.40323
PubMed Аннотация | CrossRef Полный текст | Google Scholar
51. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ и др.Картирование судьбы Т-клеток, продуцирующих ИЛ-17, при воспалительных реакциях. Nat Immunol. (2011) 12: 255–63. DOI: 10.1038 / ni.1993
PubMed Аннотация | CrossRef Полный текст | Google Scholar
52. Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO и др. TNFalpha в регуляции клеток Treg и Th27 при ревматоидном артрите и других аутоиммунных воспалительных заболеваниях. Цитокин. (2018) 101: 4–13. DOI: 10.1016 / j.cyto.2016.09.001
PubMed Аннотация | CrossRef Полный текст | Google Scholar
53.Макговерн Дж. Л., Нгуен Д. X., Нотли Калифорния, Маури К., Изенберг Д. А., Эренштейн М. Р.. Клетки Th27 сдерживаются клетками Treg посредством ингибирования интерлейкина-6 у пациентов с ревматоидным артритом, отвечающих на терапию антителами против фактора некроза опухоли. Arthritis Rheum. (2012) 64: 3129–38. DOI: 10.1002 / art.34565
PubMed Аннотация | CrossRef Полный текст | Google Scholar
54. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta в контексте воспалительной цитокиновой среды поддерживает дифференцировку de novo Т-клеток, продуцирующих IL-17. Иммунитет. (2006) 24: 179–89. DOI: 10.1016 / j.immuni.2006.01.001
PubMed Аннотация | CrossRef Полный текст | Google Scholar
55. Ивамото С., Иваи С., Цудзияма К., Курахаши К., Такешита К., Наое М. и др. TNF-альфа заставляет моноциты CD14 + человека дифференцироваться в дендритные клетки CD70 +, вызывая ответы Th2 и Th27. J Immunol. (2007) 179: 1449–57. DOI: 10.4049 / jimmunol.179.3.1449
PubMed Аннотация | CrossRef Полный текст | Google Scholar
56.Lina C, Conghua W., Nan L, Ping Z. Комбинированное лечение этанерцептом и метотрексатом меняет дисбаланс Th2 / Th3, Th27 / Treg у пациентов с ревматоидным артритом. J Clin Immunol. (2011) 31: 596–605. DOI: 10.1007 / s10875-011-9542-6
PubMed Аннотация | CrossRef Полный текст | Google Scholar
57. Талотта Р., Берзи А., Атзени Ф., Баттиччиотто А., Клеричи М., Сарци-Путтини П. и др. Парадоксальное увеличение количества лимфоцитов Th2 и Th27 при ревматоидном артрите после лечения инфликсимабом: возможное объяснение отсутствия клинического ответа. J Clin Immunol. (2015) 35: 550–7. DOI: 10.1007 / s10875-015-0182-0
PubMed Аннотация | CrossRef Полный текст | Google Scholar
58. Альзабин С., Абрахам С.М., Тахер Т.Э., Палфриман А., Халл Д., МакНами К. и др. Неполный ответ воспалительного артрита на блокаду TNF-альфа связан с путем Th27. Ann Rheum Dis. (2012) 71: 1741–8. DOI: 10.1136 / annrheumdis-2011-201024
PubMed Аннотация | CrossRef Полный текст | Google Scholar
59.Нотли, Калифорния, Инглис Дж. Дж., Альзабин С., Макканн Ф. Э., МакНэми К. Э., Уильямс РО. Блокада фактора некроза опухоли при артрите, индуцированном коллагеном, выявляет новый иммунорегуляторный путь для клеток Th2 и Th27. J Exp Med. (2008) 205: 2491–7. DOI: 10.1084 / jem.20072707
PubMed Аннотация | CrossRef Полный текст | Google Scholar
60. Wing JB, Tanaka A, Sakaguchi S. Гетерогенность и функция регуляторных Т-клеток FOXP3 (+) человека при аутоиммунитете и раке. Иммунитет. (2019) 50: 302–16.DOI: 10.1016 / j.immuni.2019.01.020
PubMed Аннотация | CrossRef Полный текст | Google Scholar
62. Беттелли Е., Носитель Y, Гао В., Корн Т., Стром Т. Б., Оукка М. и др. Взаимные пути развития для генерации патогенных эффекторных Th27 и регуляторных Т-клеток. Природа. (2006) 441: 235–8. DOI: 10.1038 / nature04753
PubMed Аннотация | CrossRef Полный текст | Google Scholar
63. Чжоу Л., Иванов И.И., Спольски Р., Мин Р., Шендеров К., Эгава Т. и др.IL-6 программирует дифференцировку клеток T (H) -17, способствуя последовательному включению путей IL-21 и IL-23. Nat Immunol. (2007) 8: 967–74. DOI: 10.1038 / ni1488
PubMed Аннотация | CrossRef Полный текст | Google Scholar
64. Корн Т., Беттелли Э., Гао В., Авасти А., Ягер А., Стром Т. Б. и др. IL-21 инициирует альтернативный путь индукции провоспалительных клеток T (H) 17. Природа. (2007) 448: 484–7. DOI: 10.1038 / nature05970
PubMed Аннотация | CrossRef Полный текст | Google Scholar
65.Самсон М., Аудиа С., Джаникашвили Н., Сьюдад М., Трад М., Фрашак Дж. И др. Краткий отчет: ингибирование функции интерлейкина-6 корректирует дисбаланс клеток Th27 / Treg у пациентов с ревматоидным артритом. Arthritis Rheum. (2012) 64: 2499–503. DOI: 10.1002 / art.34477
PubMed Аннотация | CrossRef Полный текст | Google Scholar
66. Занин-Жоров А., Динг Ю., Кумари С., Аттур М., Хиппен К. Л., Браун М. и др. Протеинкиназа С-тета опосредует отрицательную обратную связь в отношении функции регуляторных Т-клеток. Наука. (2010) 328: 372–6. DOI: 10.1126 / science.1186068
PubMed Аннотация | CrossRef Полный текст | Google Scholar
67. Занин-Жоров А., Лин Дж., Шер Дж., Кумари С., Блэр Д., Хиппен К. Л. и др. Большой гомолог 1 диска каркасного белка необходим для индуцированной Т-клеточным рецептором активации регуляторной функции Т-клеток. Proc Natl Acad Sci USA. (2012) 109: 1625–30. DOI: 10.1073 / pnas.1110120109
PubMed Аннотация | CrossRef Полный текст | Google Scholar
68.Nie H, Zheng Y, Li R, Guo TB, He D, Fang L и др. Фосфорилирование FOXP3 контролирует функцию регуляторных Т-клеток и ингибируется TNF-альфа при ревматоидном артрите. Nat Med. (2013) 19: 322–8. DOI: 10,1038 / нм.3085
PubMed Аннотация | CrossRef Полный текст | Google Scholar
69. Саломон Б.Л., Леклерк М., Тозелло Дж., Ронин Э., Пьяджио Э., Коэн Дж.Л. Фактор некроза опухоли альфа и регуляторные Т-клетки в онкоиммунологии. Front Immunol. (2018) 9: 444. DOI: 10.3389 / fimmu.2018.00444
PubMed Аннотация | CrossRef Полный текст | Google Scholar
70. Сарагоса Б., Чен Х, Оппенгейм Дж. Дж., Байенс А., Грегуар С., Чейдер Д. и др. Подавляющая активность регуляторных Т-клеток человека сохраняется в присутствии TNF. Nat Med. (2016) 22: 16–7. DOI: 10,1038 / нм 4019
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Хроническое воспаление тканей и нарушение обмена веществ
- Юн Сок Ли и
- Джеррольд Олефски
- Департамент медицины, Отдел эндокринологии и метаболизма, Калифорнийский университет в Сан-Диего, Ла-Хойя, Калифорния 92093, США
- Автор, ответственный за переписку: jolefsky {at} ucsd.edu
Аннотация
Ожирение является наиболее частой причиной инсулинорезистентности, и нынешняя эпидемия ожирения приводит к параллельному росту заболеваемость СД2. В настоящее время широко признано, что хроническое подострое воспаление тканей является основным этиологическим компонентом патогенез инсулинорезистентности и метаболической дисфункции при ожирении.Здесь мы суммируем последние достижения в нашем понимании иммунометаболизма. Мы обсуждаем характеристики хронического воспаления в основных метаболических тканях и то, как ожирение запускает эти события, включая акцент на роли гипоксии жировой ткани и экзосом, происходящих из макрофагов. Наконец, мы также рассмотреть текущие и потенциальные новые терапевтические стратегии, основанные на иммуномодуляции.
Инсулинорезистентность — ключевая особенность ожирения и сахарного диабета 2 типа (СД2).Поскольку ожирение — самая частая причина Из-за инсулинорезистентности у людей эпидемия ожирения является основной причиной роста глобальной заболеваемости СД2. У недиабетических у субъектов с ожирением гиперинсулинемия обычно компенсирует лежащую в основе инсулинорезистентность, поддерживая гомеостаз глюкозы в пределах нормального или близкого к нормальному уровню. Когда компенсаторная гиперинсулинемия не дает результатов и уровень инсулина снижается из-за развития дефекта β-клеток, гипергликемии и типичного состояния T2DM, что указывает на то, что как инсулинорезистентность, так и дисфункция β-клеток необходимы для полноценного развития этого заболевания.Известно, что хроническое воспаление тканей является ключевым фактором ожирения. к снижению чувствительности к инсулину, особенно в жировой ткани и печени. Однако несколько недавних отчетов показали воспалительные реакции островков поджелудочной железы, связанные с ожирением (Zhao et al. 2003; Warren et al. 2006; Ehses et al. 2007; Böni-Schnetzler et al. 2008; Richardson et al. 2009; Mahdi et al. 2012; Jourdan et al. 2013 г .; Камада и др. 2013 г .; Бутчер и др. 2014 г .; Хаснайн и др.2014; Ying et al. 2019), предполагая, что хроническое воспаление островков также может приводить к дисфункции β-клеток у пациентов с СД2 с ожирением.
Хотя существует ряд потенциальных, а иногда и перекрывающихся механизмов, которые могут способствовать развитию инсулинорезистентности. и дисфункция β-клеток, в этом текущем обзоре мы сосредотачиваемся на роли хронического воспаления тканей в этих метаболических дефектах. Читателя отсылают к другим отличным обзорам возможных причин инсулинорезистентности и дисфункции β-клеток, независимых от других источников. хронического воспаления тканей (Halban et al.2014; DeFronzo et al. 2015; Чешский 2017; Ньюгард 2017; Guilherme et al. 2019; Кан и др. 2019; Роден и Шульман 2019; Scherer 2019; Alonge et al. 2021; Sangwung et al. 2020).
Взаимосвязь между воспалением и метаболической дисфункцией часто описывается термином иммунометаболизм (Hotamisligil 2017). Иммунометаболизм включает влияние иммунных клеток на регуляцию системного метаболизма, а также эффекты метаболизма в иммунных клетках при воспалении.Оба эти механизма важны, но в этом обзоре мы сосредоточимся на Обращаем внимание на роль хронического воспаления в этиологии нарушения гомеостаза глюкозы.
Хроническое воспаление жировой ткани, печени, центральной нервной системы, желудочно-кишечного тракта, поджелудочной железы островки и мышцы были описаны в состояниях ожирения / СД2, что указывает на наличие сложной многоорганной сети перекрестных помех. при этих расстройствах.Общие метаболические последствия воспаления тканей, вызванного ожирением, суммированы в Таблице 1. Эти концепции в основном получены из исследований на мышах, и среди всех этих тканей, способствующих развитию, воспаление жировой ткани. при ожирении изучена наиболее полно.
Таблица 1.Метаболические эффекты воспаления тканей, связанного с ожирением
Хотя влияние хронического воспаления при СД2 на ожирение предполагалось в течение многих лет, в последние два десятилетия был опубликован ряд исследований, раскрывающих основные физиологические и молекулярные механизмы того, как ожирение связано воспаление приводит к инсулинорезистентности и непереносимости глюкозы.Об одном из самых ранних исследований сообщили Грюнфельд и Feingold (Feingold et al. 1989), который показал, что лечение грызунов TNFα приводит к гипергликемии. Дополнительное ключевое раннее исследование показало повышение TNFα уровни в жировой ткани мышей с ожирением и показали, что нейтрализация TNFα улучшает инсулинорезистентность (Hotamisligil et al. 1993). Многочисленные исследования выявили различные молекулы в каскаде воспалительных сигналов, которые могут нарушать передачу сигналов инсулина. такие как JNK, IKKβ и различные цитокины (Xia et al.2015; Goldfine and Shoelson 2017; Hotamisligil 2017; Zhao et al. 2018; Донат и др. 2019; Hall et al. 2020). Многие отчеты выявили механистическую связь между хроническим воспалением и инсулинорезистентностью в исследованиях на грызунах, проведенных использование различных генетических исследований увеличения и уменьшения функции (Lee et al. 2018). Например, мыши, созданные для глобального истощения IKKβ, JNK, CCR2 или CCL2 / MCP-1, демонстрируют повышенную чувствительность к инсулину при ожирении. (Хиросуми и др., 2002; Аркан и др.2005; Weisberg et al. 2006 г.). KO адипоцитов или гепатоцитов JNK, HIF-1α, NEMO или TLR4 также вызывают повышенную чувствительность к инсулину (Sabio et al. 2008; Wunderlich et al. 2008; Jia et al. 2014; Lee et al. 2014). Напротив, адипоцит-специфическая сверхэкспрессия Ccl2 или Hif1a вызывает инсулинорезистентность и непереносимость глюкозы при нормальной диете (Kanda et al. 2006; Halberg et al. 2009). Наиболее важно то, что большое количество специфичных для макрофагов (или гемопоэтических клеток) KO воспалительных молекул было обнаружено. сообщили, что все они приводят к защите от инсулинорезистентности и непереносимости глюкозы (Saberi et al.2009; Олефский и Гласс 2010; Han et al. 2013; Hill et al. 2014; Takikawa et al. 2016; Desai et al. 2017). Однако, несмотря на то, что существует ряд исследований жировой ткани человека (Lofgren et al. 2000; Fabbrini et al. 2013; Hill et al. 2018), печень (Senn et al. 2002; Ghazarian et al. 2017) является первичной адипоциты (Liu et al. 1998), скелетные мышцы (Austin et al. 2008) и островки поджелудочной железы (Maedler et al. 2002), которые поддерживают этот процесс иммунометаболизма (Pickup et al. 1997), концепция, согласно которой хроническое воспаление вызывает инсулинорезистентность и дисфункция β-клеток еще предстоит подтвердить в люди.
Связь между СД2 и генетическими вариациями, связанными с воспалением
Хотя вклад хронического воспаления в развитие инсулинорезистентности и СД2 еще предстоит подтвердить в у людей, недавние достижения в исследованиях полногеномных ассоциаций (GWAS) предполагают, что воспаление может быть причинно связано с заболеваемость СД2 у людей.В то время как ранние исследования GWAS показали, что СД2 связан с генетическими вариациями в генах, связанных с с секрецией инсулина и функцией β-клеток (McCarthy et al. 2010; Dimas et al. 2014), более поздние исследования показали, что гены, участвующие в периферической чувствительности к инсулину и функции жировой ткани, такие как PPARG и KLF14 , также связаны с заболеваемость СД2 (Войт и др., 2010). Более того, вариации генов, регулирующих Т-клетки (например, PTPRJ и CMIP ) или макрофаги (например, PTPRJ и CMIP ) или макрофаги.g., MAEA ) или воспалительные сигнальные пути (например, WWOX , MAP8IP1 , IFNGR1 , ST6GAL1 , JAZF1 , MAP3K1 , TLR и MACROD1 ) также связаны с заболеваемостью СД2 (Waeber et al. 2000; Kooner et al. 2011; Cho et al. 2012; Manning et al. 2012; Locke et al. 2015; Shungin et al. 2015; Flannick et al. . 2019; Liao et al.2019; Diedisheim et al.2020).В то время как обновляются более инклюзивные исследования GWAS и полного экзома (Mahajan et al., 2018; Flannick et al., 2019), многие генетические вариации, связанные с заболеваемостью T2DM, обнаруживаются в некодирующих областях ДНК. Более того, неоднородность в этиологии СД2 (Philipson 2020) может снизить влияние конкретных генетических вариаций на патологию заболевания. Следует учитывать эти факторы. при интерпретации результатов GWAS.
Воспаление при ожирении
Жировая ткань
Хроническое воспаление жировой ткани является характерной чертой ожирения, и одновременно были опубликованы два ранних исследования. в 2003 г. (Weisberg et al.2003; Xu et al. 2003) показали, что это во многом связано с повышенным накоплением провоспалительных макрофагов в жировой ткани человека и мыши. ткань. Многие последующие отчеты задокументировали это накопление макрофагов жировой ткани (АТМ) в жировой ткани с ожирением. и дополнительно охарактеризовали эти макрофаги в отношении поверхностных маркеров, биологических эффектов и транскриптомных фенотипов. Помимо макрофагов, в воспалении жировой ткани с ожирением участвуют другие типы клеток врожденного и адаптивного иммунитета (резюмировано на рис.1). Однако макрофаги являются доминирующим типом клеток, опосредующим инсулинорезистентность и метаболическую дисфункцию при ожирении. ткань. Таким образом, макрофаги являются наиболее распространенным типом иммунных клеток, накапливающихся в жировой ткани и печени, страдающих ожирением. Кроме того, эти клетки являются основными иммунными клетками, секретирующими большинство воспалительных цитокинов, галектина-3 и экзосом (Weisberg et al. 2003; Zeyda et al. 2007; Li et al. 2016; Ying et al. 2017a), а также истощение макрофагов. корректирует инсулинорезистентность и толерантность к глюкозе у мышей с ожирением (Huang et al.2010; Chatenoud et al. 2011; Ли и др. 2011b; Bu et al. 2013).
Рисунок 1.Воспаление жировой ткани при ожирении. В нормальном состоянии постоянно действующие банкоматы демонстрируют M2-подобный поляризованный фенотип. Факторы высвобождаемые из Tregs и эозинофилов поддерживают банкоматы для поддержания этого противовоспалительного состояния. При ожирении увеличиваются адипоциты Продукция хемокинов вызывает повышенное рекрутирование моноцитов крови, а также пролиферацию ATM.Большинство моноцитарных производных ATM экспрессируют CD11c и / или CD9 и M1-подобный поляризованный фенотип. Уменьшение количества эозинофилов и Treg и увеличение количество нейтрофилов, ILC1, CD8 + T-клетки, Th2-клетки и B2-клетки усиливают M1-подобную поляризацию ATM и воспаление жировой ткани.
Макрофаги жировой ткани
У худых мышей макрофаги составляют 5–10% стромальных сосудистых клеток висцеральной жировой ткани.Уже через 3 дня после кормления диета с высоким содержанием жиров (HFD), повышенная экспрессия провоспалительных цитокинов и инфильтрация макрофагов могут наблюдаться при висцеральном жировая ткань (Lee et al. 2011b). Эти изменения постепенно усиливаются по мере развития ожирения вместе с прогрессирующим ухудшением чувствительности к инсулину. и толерантность к глюкозе, пока ожирение полностью не стабилизируется и эти изменения не начнут выходить на плато. При ожирении банкоматы накапливаются, становясь наиболее распространенный тип иммунных клеток в жировой ткани и может составлять до 40% всех стромальных сосудистых клеток.Вместе с количественные изменения в содержании ATM, вызванные ожирением, ATM также претерпевают фенотипическое переключение с M2-подобных поляризованных противовоспалительных клетки к M1-подобному поляризованному провоспалительному состоянию (Lumeng et al. 2007a). Считается, что макрофаги M1 представляют «классически активированный» провоспалительный фенотип макрофагов, и эти макрофаги производят провоспалительные цитокины, активные формы кислорода и оксид азота, которые устраняют патогены.М2 клетки представляют собой «альтернативно активированный» противовоспалительный фенотип макрофагов, которые обычно способствуют восстановлению тканей и ремоделирование (Jakubzick et al., 2017; Koelwyn et al., 2018). При ожирении, в то время как количество как M1-, так и M2-подобных поляризованных банкоматов увеличивается, увеличение количества M1-подобных поляризованных банкоматов намного больше, смещая баланс в сторону провоспалительного состояния ткани. Большинство M1-подобных поляризованных банкоматов являются производными из циркулирующих моноцитов и экспрессируют CD11c на поверхности, тогда как большинство M2-подобных поляризованных ATM являются резидентными и не отображают экспрессию CD11c (Lumeng et al.2007b). Воспалительное состояние банкоматов также может быть изменено различными видами липидов. Насыщенные жирные кислоты (НЖК), полученные из либо диетические источники, либо гидролиз триглицеридов адипоцитов могут управлять M1-подобной поляризацией ATM через toll-подобный рецептор 4 (TLR4) -зависимый механизм (Saberi et al. 2009; Holland et al. 2011; Orr et al. 2012; Tao et al. 2017). С другой стороны, жирные кислоты омега-3 ингибируют провоспалительную активацию ATM посредством GPR120-зависимого механизма (Oh et al.2010).
Термины M1- и M2-like часто используются для описания банкоматов в худых и страдающих ожирением состояниях для удобства. Тем не менее концепция макрофагов M1 и M2 была впервые введена на основании иммунологических различий между линиями мышей, которые преимущественно продвигать ответы Th2 или Th3. Их конкретно определяют как клетки-предшественники костного мозга, поляризованные до состояния M1 или M2. в культуре путем инкубации с LPS / IFNγ или IL-4 / IL-13 соответственно (Mills et al.2000). В условиях in vivo банкоматы не существуют в рамках этих узких категорий, но фактически охватывают весь спектр поляризации. похожи на макрофаги M1 или M2, но отличаются от них (Li et al., 2019). Таким образом, исследования с использованием анализа секвенирования одноклеточной РНК (scRNA-seq) показали, что транскриптомные профили ATM сильно неоднороден в ожирении жировой ткани. Банкоматы при ожирении демонстрируют провоспалительный профиль, который перекрывается, но отличается по сравнению с клетками M1, полученными in vitro (Kratz et al.2014; Hill et al. 2018; Jaitin et al. 2019). Более того, они демонстрируют уникальные профили экспрессии метаболических генов, включая активацию лизосомно-зависимого липидного метаболизма, вместе называемое «состояние метаболической активации» (Xu et al. 2013; Kratz et al. 2014; Coats et al. 2017). Hill et al. (2018) сообщили о двух субпопуляциях ATM в зависимости от экспрессии CD9, и количество ATM CD9 + увеличивается во время развития ожирения. Эти CD9 + ATM демонстрируют уникальные функциональные, морфологические и локальные особенности: они экспрессируют высокие уровни провоспалительных цитокинов, содержат внутриклеточные липидные капли, секретируют экзосомы и расположены внутри короноподобных структур, окружающих умирающие адипоциты.Кроме того, подмножество ATM экспрессирует маркер моноцитов Ly6C, а также накапливается в жировой ткани с ожирением. Начиная с Ly6C является маркером для моноцитов, Ly6C + ATM, скорее всего, представляют недавно рекрутированные макрофаги, происходящие из моноцитов, подвергающиеся дифференцировке. Как обнаружено в стенке аорты макрофаги, эти клетки Ly6C + теряют экспрессию Ly6C, поскольку они полностью дифференцируются в зрелые макрофаги (Jakubzick et al.2017). Ly6C + ATM находятся за пределами короноподобных структур и могут стимулировать дифференцировку адипоцитов (Hill et al. 2018). Многие из генов транскриптомных сигнатур ATM CD9 + или Ly6C + отличаются от дифференцированных in vitro макрофагов M1 или M2 или друг от друга. Jaitin et al. (2019) провели объективный анализ scRNA-seq и обнаружили, что банкоматы можно разделить на три субпопуляции: одна демонстрирует транскриптомную сигнатуры периваскулярных макрофагов и еще двух подгрупп с экспрессией CD9.В соответствии с выводами Hill et al. (2018) они сообщили, что количество банкоматов CD9 + увеличивается при ожирении и может быть обнаружено внутри короноподобных структур. Они дополнительно охарактеризовали CD9 + ATM с использованием отслеживания клонов и обнаружили, что ~ 80% ATM CD9 + происходят из костного мозга. Интересно, что ATM CD9 + можно разделить на две субпопуляции в зависимости от экспрессии липидного рецептора Trem2. Trem2 + CD9 + ATM в большом количестве экспрессируют гены, связанные с фагоцитозом и метаболизмом липидов.Они назвали эти клетки липид-ассоциированными. макрофагов (LAM) и показали, что экспансия LAM, вызванная ожирением, зависит от TREM2 и что мыши Trem2 KO демонстрируют повышенный набор веса, непереносимость глюкозы и дислипидемию на HFD, что позволяет предположить, что TREM2-зависимые LAM ATM может уменьшить ожирение и нарушение регуляции обмена веществ.
Эти CD9 + Trem2 + ATM демонстрируют функциональное сходство с CD11c + , M1-подобными поляризованными ATM, а экспрессия CD9 коррелирует с экспрессией CD11c, хотя популяции CD11c + и CD9 + не идентичны (Hill и другие.2018). Это предполагает, что популяции CD9 + и CD11c + могут быть функционально гетерогенными. Требуются дополнительные исследования для дальнейшего определения двойных и двойных CD11c и CD9. единичные положительные и отрицательные популяции.
Банкоматы и контроль местного уровня катехоламинов
Чавла и его коллеги предположили, что M2-подобные поляризованные ATM синтезируют катехоламины (Nguyen et al.2011; Qiu et al. 2014). Однако эта концепция была оспорена другими группами, которые сообщили, что банкоматы не производят физиологически значимых уровней. катехоламинов и не экспрессируют фермент тирозингидроксилазу, который необходим для синтеза катехоламинов. Кроме того, эти группы обнаружили, что лечение ИЛ-4 не влияет на расход энергии и массу тела ни на холоде, ни в окружающей среде, ни при термонейтральном воздействии. температуры (Fischer et al.2017). В качестве альтернативного механизма того, как макрофаги могут модулировать локальные уровни катехоламинов, подмножество макрофагов, называемое Макрофаги, связанные с симпатическими нейронами (SAM), способны к внеклеточному захвату катехоламинов и катаболизму (Pirzgalska et al.2017). SAM представляют собой другое подмножество ATM, обнаруживаемое вблизи симпатических нервов в жировой ткани. При ожирении количество SAMs увеличивается, что способствует снижению адаптивной термогенной активности.Концепция катехоламинов, опосредованных макрофагами О катаболизме в жировой ткани также сообщили Диксит и его коллеги (Camell et al.2017), которые показали, что резистентность к катехоламинам, вызванная старением, связана с повышенным катаболизмом катехоламинов ATM. В поддержку концепции о том, что банкоматы регулируют локальные уровни катехоламинов, системное истощение тканевых макрофагов с помощью липосом клодроната, но не симпатическая денервация, блокирует образование паховой WAT у мышей с адипоцитами Fasn KO (Henriques et al.2020).
Механизмы накопления макрофагов
Механизм, с помощью которого макрофаги накапливаются в жировой ткани с ожирением, включает (1) усиление хемотаксиса моноцитов / макрофагов. (увеличение иммиграции моноцитов крови) и снижение эмиграции из банкоматов (или увеличение удержания), и (2) более высокий уровень резидента Распространение банкоматов.Мечение in vivo моноцитов крови или резидентных банкоматов показало, что миграция моноцитов крови в жировую ткань ткани увеличивается при ожирении (Lumeng et al. 2007a; Oh et al. 2012). Это увеличение связано с моноцитозом (Nagareddy et al. 2014) и повышенной продукцией хемокинов, таких как CCL2 / MCP-1 и лейкотриен B4 (LTB4), в висцеральной жировой ткани (Li et al. 2015). Специфическое для адипоцитов истощение CCL2 снижает вызванное ожирением накопление ATM (Kanda et al. 2006). Более того, генетическая делеция или фармакологическое ингибирование рецепторов хемокинов CCL2 (CCR2) или LTB4 (LTB4R) блокирует накопление АТМ, вызванное ожирением (Lumeng et al.2007a; Ли и др. 2015). Это показывает, что хемотаксис моноцитов в жировую ткань в значительной степени опосредуется системами CCL2 / CCR2 и LTB4 / LTB4R. В Помимо повышенного хемотаксиса, уровни факторов, связанных с удержанием макрофагов, таких как Netrin1 и Sema3E, увеличиваются в жировой ткани с ожирением, что препятствует эмиграции банкоматов (Shimizu et al. 2013; Wanschel et al. 2013; Ramkhelawon et al. 2014).
Помимо хемотаксиса в жировой ткани, накопление ATM также происходит из-за повышенной пролиферации резидентных ATM, которые может стимулироваться CCL2 (Amano et al.2014). В последующих исследованиях повышенная пролиферация ATM в жировой ткани с ожирением была подтверждена отдельными группами (Zheng et al., 2016).
Другие клетки врожденного иммунитета
Несмотря на то, что их количество относительно невелико, изменения в других типах клеток врожденного иммунитета также способствуют развитию жировой ткани. воспаление тканей, в основном за счет регуляции поляризации и функции ATM.Например, клетки врожденных лимфоидных клеток 1 типа (ILC1), включая естественные клетки-киллеры, находятся в нормальной / тощей жировой ткани и способствуют гомеостазу жировой ткани (Lee et al., 2016; Boulenouar et al., 2017; Theurich et al., 2017). При ожирении количество клеток ILC1 увеличивается, а цитокины, производные от ILC1, включая IFNγ, стимулируют провоспалительную активацию. банкоматов. С другой стороны, количество клеток ILC2, продуцирующих цитокины Th3, уменьшается при ожирении (Molofsky et al.2013; Nussbaum et al. 2013). Цитокины, производные от ILC2, поддерживают иммиграцию и созревание эозинофилов в жировой ткани, которые продуцируют IL-4 и IL-13. которые стимулируют поляризацию M2-подобных макрофагов (Wu et al. 2011). В жировой ткани с ожирением уменьшение количества клеток ILC2 связано со снижением количества местных эозинофилов. Наконец, набор нейтрофилов в жировую ткань увеличивается при ожирении, а факторы, выделяемые нейтрофильными гранулами, такие как нейтрофилы эластаза и миелопероксидаза могут способствовать развитию инсулинорезистентности (Talukdar et al.2012; Tam et al. 2020).
Клетки адаптивного иммунитета
Несколько типов адаптивных иммунных клеток также вовлечены в воспаление жировой ткани. При ожирении количество жировой CD8 + в тканях Т-клеток увеличивается, и факторы, высвобождаемые из этих клеток, могут способствовать дифференцировке ATM и хемотаксису (Nishimura et al. 2009). Среди CD4 + Т-клеток количество провоспалительных Т-хелперных (Th) 1-клеток увеличивается (Nishimura et al.2009), тогда как содержание противовоспалительных клеток Th3 в жировой ткани уменьшается при ожирении, что способствует увеличению жировой массы. уровни тканевого интерферона γ (IFNγ) и воспаление (Winer et al. 2009). Tregs (CD3 + CD4 + FOXP3 + ) — еще одна субпопуляция Т-лимфоцитов CD4 + , хорошо изученная в жировой ткани (Feuerer et al. 2009; Li et al.2020). У молодых мышей имеется лишь небольшое количество этих клеток, но они начинают накапливаться во висцеральной жировой ткани в более раннем возрасте. возраст 10–15 недель из-за повышенной пролиферации и снижения оборачиваемости.Tregs представляют собой основную подгруппу CD4 + Т-клеток (40–80% от общего количества CD4 + Т-клеток) в возрасте 20–30 недель у нормальных мышей (Bapat et al. 2015). Tregs контролируют активность других Т-клеток и сдерживают врожденную иммунную систему, подавляя иммиграцию моноцитов и провоспалительная поляризация. При ожирении количество или пропорция Treg-клеток жировой ткани уменьшается, что способствует к усилению воспаления жировой ткани (Feuerer et al.2009 г.). Treg жировой ткани демонстрируют уникальный транскриптомный профиль, отличный от Treg в других сайтах, который в значительной степени регулируется рецептором γ, активируемым пролифератором пероксисом (PPARγ). Действительно,> 80% Treg жировой ткани экспрессируют PPARγ, что является уникальным к жировым Treg, а Treg PPARγ опосредует компонент противовоспалительного действия тиазолидиндионов (TZD) при ожирении мышей (Cipolletta et al. 2012). γδ Т-клетки, присутствующие в жировой ткани нормальных мышей, поддерживают симпатическую иннервацию и расширение Treg, высвобождая IL-17. (Кольгрубер и др.2018; Hu et al. 2020).
Инвариантные естественные Т-клетки-киллеры (iNKT) составляют 10–30% всех Т-клеток, находящихся в жировой ткани (Huh et al. 2013). Это врожденные αβ Т-клетки, которые специфически стимулируются липидными антигенами за счет основной гистосовместимости. комплексная (MHC) -подобная молекула CD1d (Huh et al.2013; Lynch 2014). Клетки iNKT высвобождают IL-10, IL-4, IL13 и IL-2, усиливая M2-подобную поляризацию ATM.Количество жировой ткани Количество клеток iNKT уменьшается при ожирении, и это может способствовать развитию воспаления. Однако в других отчетах сомневаюсь в благотворном влиянии клеток iNKT. Например, Satoh et al. (2016) и Wu et al. (2012b) показали провоспалительные и отрицательные метаболические эффекты iNKT-клеток у мышей с ожирением. В качестве возможного объяснения этих противоречащие результатам, анализ scRNA-seq показал, что iNKT-клетки жировой ткани можно функционально разделить на два подмножества: в зависимости от НК1.1: NK1.1 + iNKT являются провоспалительными и продуцируют IFNγ, тогда как NK1.1 — iNKT являются противовоспалительными и продуцируют IL-10 (LaMarche et al.2020).
В-лимфоциты составляют 3–10% ВПВ в нормальной жировой ткани и увеличиваются в два раза при ожирении или в 10 раз в пожилом возрасте. мыши (Camell et al., 2019). По сравнению с селезенкой и костным мозгом В-клетки жировой ткани показывают более высокое соотношение B1: B2 у нормальных мышей.Однако в При ожирении соотношение B1: B2 уменьшается с увеличением рекрутирования провоспалительных клеток B2 (Ying et al., 2017b). Рекрутирование B2 в висцеральную жировую ткань при ожирении в основном опосредуется системой LTB4 / LTB4R1 (Ying et al., 2017b; Srikakulapu and McNamara 2020), и эти клетки могут способствовать воспалению жировой ткани и метаболической дисфункции при ожирении.
Острое воспаление в сравнении с хроническим
Хотя многочисленные исследования показали пагубное влияние хронического воспаления жировой ткани на развитие системных инсулинорезистентность при ожирении, воспалении — это физиологический процесс, необходимый для защиты от патогенной инвазии или повреждение и восстановление тканей.Острое воспаление необходимо для поддержания нормальной функции жировой ткани за счет здорового ремоделирование жировой ткани для безопасного размещения избыточных липидов в жировой ткани (Wernstedt Asterholm et al. 2014). Например, Zhu et al. (2020) сообщили, что ингибирование воспалительных путей за счет индуцируемой сверхэкспрессии аденовирусного белка, называемого RIDα / β (который блокирует несколько воспалительных сигнальных путей) снижает прибавку в весе и ожирение и усугубляет непереносимость глюкозы и инсулина у мышей HFD / ожирения.Следовательно, вполне вероятно, что острое воспаление жировой ткани необходимо для гомеостаза жировой ткани. тогда как хроническое воспаление способствует развитию инсулинорезистентности и дисфункции жировой ткани.
Печень
Печеночные макрофаги
Ожирение, или HFD, приводит к стеатозу печени, обычно называемому неалкогольной жировой болезнью печени (НАЖБП), которая может перейти к состоянию, называемому неалкогольным стеатогепатитом (НАСГ).Ожирение также связано с усилением воспаления печени, а макрофаги являются основным источником провоспалительных цитокинов (Stienstra et al. 2010; Xu et al. 2014). Инъекция липосом гадолиния или клодроната внутривенно избирательно истощает печеночные макрофаги из-за фенестрированных печеночная синусоидальная структура. Избирательное истощение печеночных макрофагов улучшает инсулинорезистентность и стеатоз печени у тучные мыши или крысы (Huang et al.2010; Lanthier et al. 2010; Ли и др. 2011b; Reid et al. 2016). Макрофаги печени можно разделить на две основные подгруппы в зависимости от их происхождения: они включают происходящие из желточного мешка. резидентные клетки Купфера (KC) и происходящие из костного мозга макрофаги печени (RHM). В нормальной печени печеночные макрофаги составляют ∼10% всех печеночных клеток, и большинство из них являются KCs (Bouwens et al. 1986). При ожирении количество KC не изменяется, тогда как хемотаксис циркулирующих моноцитов и дифференцировка в макрофаги (RHM) увеличиваются, в основном за счет системы CCL2 / CCR2 (Obstfeld et al.2010; Morinaga et al. 2015). Хотя и KC, и RHM очень гетерогены, RHM экспрессируют более высокие уровни M1-подобных поляризованных маркеров макрофагов и экспрессия провоспалительных генов, которая усугубляется при ожирении (Arkan et al. 2005; Morinaga et al. 2015; Seidman et al. 2020). Эти результаты предполагают, что основной компонент воспаления печени, вызванного ожирением, может быть связан с увеличением количества моноцитов. инфильтрация и дифференцировка макрофагов (суммированные на рис.2).
Фигура 2.Воспаление печени. В нормальном физиологическом состоянии KC составляют ~ 10% всех клеток печени. Кроме того, они собирают мусор патогены и демонстрируют M2-подобный поляризованный противовоспалительный фенотип. В печени с ожирением и стеатозом KC могут проявлять повышенную экспрессию. генов, связанных с восстановлением тканей и воспалением.Эти гены включают Cd9 и Trem2 . Это сопровождается повышенным апоптозом KC, и компонент гибели KC компенсируется повышенным рекрутированием моноциты крови, которые дифференцируются в KC-подобные макрофаги. Хемокины, выделяемые стеатотическими гепатоцитами, вызывают повышенное рекрутирование моноцитов крови в печень, которые дифференцируются в провоспалительные макрофаги (RHM) и продуцируют факторы что может вызвать инсулинорезистентность.Набор нейтрофилов также увеличивается, и молекулы, высвобождаемые из нейтрофилов, гранулы, такие как эластаза нейтрофилов и миелопероксидаза, могут вызывать инсулинорезистентность в гепатоцитах.
Недавно проведен одноклеточный анализ транскриптомных и эпигенетических изменений печеночных макрофагов (например, scRNA-seq и scATAC-seq) в сочетании с отслеживанием моноцитов и исследованиями истощения KC дополнительно выяснились фенотипы печеночных макрофагов (Seidman et al.2020). Таким образом, у мышей, которых кормили HFD с предрасположенностью к НАСГ, KC частично теряют характерные для них паттерны экспрессии генов, в то время как фенотип связанных с рубцами макрофагов с экспрессией Trem2 и Cd9 . Эти изменения сопровождаются усилением апоптоза KC. По мере того, как эти числа KC снижаются, они заменяются моноцитарными макрофаги, которые претерпевают конвергентные эпигеномные и транскриптомные изменения и становятся новыми KC-подобными клетками.
Другие иммунные клетки печени
Количество нейтрофилов также увеличивается в печени мышей с ожирением по сравнению с худыми / нормальными мышами (Talukdar et al. 2012), а цитокины и хемокины, полученные из нейтрофилов, способствуют развитию воспаления печени, вызванного ожирением. Кроме того, NE, высвобождаемый из нейтрофилов, может захватываться гепатоцитами и вызывать деградацию IRS-2, способствуя снижению чувствительности к инсулину.
Среди компонентов адаптивного иммунитета количество цитотоксических Т-клеток CD8 + увеличивается в печени мышей с ожирением по сравнению с худыми мышами через IFNγ-зависимый сигнальный путь (Газарян и др., 2017). С другой стороны, количество CD4 + T-клеток, Treg и γδ T-клеток уменьшается или остается неизменным при ожирении.
Связь между типами клеток печени
Вызванные ожирением изменения в печени, ведущие к развитию инсулинорезистентности и НАСГ, связаны с взаимодействием между типы внутрипеченочных клеток, такие как гепатоциты, различные иммуноциты и звездчатые клетки печени (Koyama and Brenner 2017).Например, активация гепатоцитов JNK, IKKβ или IKKε или ответ развернутого белка (стресс ER) увеличивает продукцию хемокинов и других гепатокинов, стимулирующих провоспалительную активацию KC и набор циркулирующих моноцитов и нейтрофилов (Аркан и др., 2005; Секи и др., 2012; Рейли и др., 2013; Ли и др., 2018). Поступающие иммунные клетки обладают сильным провоспалительным действием (Morinaga et al. 2015; Seidman et al. 2020), усиливая воспаление печени.Факторы, происходящие из гепатоцитов и иммуноцитов, такие как TGFβ, могут активировать покоящиеся звездчатые клетки. и вызывают секрецию коллагена, что приводит к фиброзу печени (Hellerbrand et al. 1999; Koyama and Brenner 2017). Более того, активированные цитокины и хемокины звездчатых клеток могут усиливать воспаление печени, усугубляя стеатоз печени. и фиброз (Weiskirchen and Tacke 2014). См. Koyama and Brenner (2017) и Arab et al. (2018) за прекрасные обзоры по патофизиологии НАЖБП и НАСГ.
Мышцы
На скелетные мышцы приходится большая часть стимулированного инсулином удаления глюкозы in vivo, и это количество снижается при ожирении / СД2, способствующие развитию компенсаторной гиперинсулинемии и непереносимости глюкозы. Несколько механизмов, таких как митохондриальные дисфункция, микрососудистая дисфункция и эктопическое накопление жира (или липотоксичность) были предложены в качестве причин этого дефект и хорошо рассмотрены в других источниках (Serné et al.2007; Роден и Шульман 2019; Sergi et al. 2019). Хотя они относительно менее изучены по сравнению с жировой тканью и печенью, провоспалительные пути усиливаются с увеличением инфильтрация макрофагов в скелетных мышцах мышей с ожирением и пациентов с СД2 по сравнению с нормальными мышами или людьми (Fink et al. 2014). Системное истощение макрофагов или генетических модификаций, подавляющих провоспалительную активацию макрофагов, улучшается Чувствительность к инсулину в скелетных мышцах мышей (Patsouris et al.2008; Han et al. 2013), предполагая, что воспаление играет причинную роль в развитии мышечной инсулинорезистентности. В соответствии с этим, лечение провоспалительными цитокинами вызывает инсулинорезистентность в культивируемых миоцитах или скелетных мышцах in vivo (de Alvaro et al. 2004; Plomgaard et al. 2005). С другой стороны, противовоспалительный цитокин IL-10 может играть защитную роль. Например, мышечно-специфическая сверхэкспрессия IL-10 смягчает, а специфическая для мышц делеция Il10 усугубляет вызванное ожирением воспаление мышц и инсулинорезистентность (Hong et al.2009; Дагдевирен и др. 2016).
Что касается механизмов воспаления мышц, межмиоцеллюлярной жировой ткани (IMAT) или перимускулярной жировой ткани (PMAT) были замешаны (Хан и др., 2015). Таким образом, при ожирении IMAT и PMAT расширяются и демонстрируют накопление макрофагов, подобное висцеральной жировой ткани. Как в висцеральном жировой ткани, большинство макрофагов, инфильтрированных в скелетные мышцы с ожирением, представляют собой CD11c + M1-подобные поляризованные макрофаги.Более того, изменения в доле различных подмножеств Т-клеток (например, увеличение количества клеток Th2 и уменьшенные Treg) демонстрируют те же закономерности, что и в висцеральной жировой ткани (Wu and Ballantyne, 2017).
Островки поджелудочной железы
Дисфункция β-клеток является критическим этиологическим компонентом СД2, и были идентифицированы различные механизмы этого дефекта. (обзоры см. в Halban et al.2014; Hudish et al. 2019). Несколько отчетов показали, что воспаление островков также может быть этиологически важным в возникновении дисфункции β-клеток при СД2. Экспрессия провоспалительных цитокинов (таких как IL-1β, IL-33, IL-23 и IL-24) и инфильтрация макрофагами увеличивается. в островках пациентов с ожирением / СД2 по сравнению с нормальными субъектами (Ehses et al. 2007; Böni-Schnetzler et al. 2008; Richardson et al. 2009; Mahdi et al. 2012; Butcher et al. 2014; Hasnain et al.2014; Kamata et al. 2014; Schludi et al. 2017). Обработка изолированных островков человека провоспалительными цитокинами снижает индуцированные глюкозой внутриклеточные уровни Ca 2+ и GSIS (Corbett et al. 1993; O’Neill et al. 2013) и индуцирует гибель β-клеток (Brozzi et al. 2015) . В отличие от островков СД1, в воспалении островков, связанных с ожирением или СД2, преобладают макрофаги и не вовлекаются Т-клетки. инфильтрация (Ehses et al. 2007). Концепция о том, что воспаление островков вызывает дисфункцию β-клеток, была более тщательно проверена в исследованиях на грызунах.Системный истощение макрофагов путем внутрибрюшинной инъекции липосом клодроната усиливает секрецию инсулина, стимулированную глюкозой (GSIS), а также чувствительность к инсулину у мышей с ожирением (Eguchi et al. 2012). Истощение ex vivo макрофагов в первичных островках, выделенных от мышей HFD / ожирения, усиливает GSIS (Eguchi et al. 2012; Ying et al. 2019). Обработка первичных островков мыши провоспалительными цитокинами (например, IL-1β, IL-23, IL-24) снижает GSIS (Mahdi et al.2012; Jourdan et al. 2013; Hasnain et al. 2014). Более того, мыши Myd88 или TLR4 KO защищены от дисфункции β-клеток, вызванной субхронической инфузией липидов (липотоксичность). (Eguchi et al. 2012), в то время как у мышей KO, специфичных для β-клеток, антагонистов рецепторов IL-1 (IL-1Ra) развивается β-клеточная дисфункция (Böni-Schnetzler et al. 2018).
Хотя аналогично воспалению жировой ткани и печени в отношении функциональных дефектов паренхиматозных клеток (в данном случае β клеток), наблюдается резкое различие в механизме накопления макрофагов в островках ожирения по сравнению с печенью и жировой тканью. ткань.Таким образом, в печени и жировой ткани распространение макрофагов в значительной степени связано с привлечением циркулирующих моноцитов. (Weisberg et al. 2003; Morinaga et al. 2015; Jaitin et al. 2019; Seidman et al. 2020a) с меньшим компонентом из-за пролиферации резидентных макрофагов (Amano et al. 2014). В островках накопление макрофагов в значительной степени опосредуется пролиферацией резидентных островковых макрофагов (Ying et al.2019). Моноциты крови рекрутируются в область периостровок, но не проникают в капсулу островка (Eguchi et al.2012; Ying et al. 2019).
В островках есть две резидентные популяции макрофагов (внутриостровковые и периостровковые макрофаги) с разными анатомическими особенностями. распределение, фенотипы и функциональные свойства (Ying et al.2019). Эти островковые макрофаги демонстрируют уникальный транскриптомный профиль, отличный от M1- или M2-подобных поляризованных макрофагов или других тканевые макрофаги. Внутриостровковые макрофаги существенно размножаются при ожирении, в то время как околостровковые макрофаги выстилаются островковая капсула — нет.Более того, внутриостровковые макрофаги, но не околостровковые макрофаги, ингибируют секрецию инсулина β-клетками. Этот эффект внутриостровных макрофагов опосредован межклеточным контактно-зависимым механизмом, включающим поглощение Везикулы секреции инсулина β-клеток. Кроме того, провоспалительные цитокины, продуцируемые островными макрофагами, также подавляют β-клетки. GSIS (Maedler et al. 2002; Eguchi et al. 2012). Например, IL-1β, продуцируемый островковыми макрофагами, связывается с рецептором IL-1 (IL-1R), который обильно экспрессируется на β-клеток, что приводит к снижению GSIS за счет стимуляции сигнальных путей JNK и NF-κB (Kwon et al.1998; Ларсен и др. 2005; Ортис и др. 2006; Wang et al. 2009 г.). Кроме того, островковые макрофаги (как внутри островка, так и вокруг островка) высвобождают факторы роста, такие как PDGF и IGF-1, имитируя пролиферация β-клеток (Ying et al.2019; Nackiewicz et al.2020). Следовательно, вероятно, что воспаление островков, связанное с ожирением, вызывает снижение GSIS β-клеток и способствует повышению β-клеток. распространение, которое существует в островках ожирения. Это приводит к изменениям функции островков, способствующим увеличению секреции базального инсулина. и уменьшил GSIS.
Можно задаться вопросом, обязательно ли повышенная пролиферация β-клеток островковыми макрофагами пагубна для метаболического гомеостаза. Действительно, хотя побочные эффекты воспаления островков на GSIS и β-функцию были зарегистрированы при хроническом ожирении и воспалительном процессе. состояний, воспаление — это физиологический процесс, и острое воспаление островков может быть полезным для поддержания островковых функционируют в нормальном состоянии (Banaei-Bouchareb et al.2004; Geutskens et al. 2005; Яно и др. 2007; Kayali et al. 2012; Ли и др. 2013; Dror et al. 2017; Burke et al. 2018; Riopel et al. 2018; Ying et al. 2019). Следовательно, возможно, что острое воспаление необходимо для поддержания функции островков, тогда как хроническое воспаление способствует развитию дисфункции β-клеток.
ЖКТ
Недавние достижения в нашем понимании микробиоты кишечника позволили по-новому взглянуть на то, как ожирение влияет на воспаление тканей и нарушения обмена веществ.При ожирении как у человека, так и у грызунов меняется состав кишечной микробиоты. (дисбактериоз), которые могут изменять иммунную систему и метаболизм хозяина. У HFD / тучных мышей WT наблюдается повышенный провоспалительный цитокин экспрессия и продуцирующие IL17 γδ Т-клетки, Th2-клетки, CD8 + Т-клетки, макрофаги и дендритные клетки, а также сниженные Treg, клетки ILC3 и эозинофилы в кишечной пластине propria (Джонсон и др.2015; Монтейро-Сепульведа и др. 2015; Winer et al. 2016). Эксперименты по трансплантации фекалий и клинические испытания также показали, что дисбактериоз способствует развитию воспаления. и инсулинорезистентность при ожирении. Например, трансплантация стерильных мышей с фекальными препаратами от человеческих близнецов. дискордантный для ожирения показал, что микробиота кишечника с ожирением может вызывать увеличение веса, массы жировой ткани и глюкозы. непереносимость (Ridaura et al.2013). Более того, клиническое исследование показало, что введение кишечной микробиоты от худых доноров реципиентам с метаболическим синдром повышенной чувствительности к инсулину (Vrieze et al. 2012). Эти исследования предполагают, что компонент метаболического фенотипа, связанного с ожирением, является следствием дисбактериоза. Тем не менее, Следует отметить, что эффект от трансплантации фекалий был небольшим, а перенос здорового кишечного микробиома не нормализовался. метаболические нарушения, вызванные ожирением.
В этом контексте несколько молекул / механизмов кишечной микробиоты, которые могут изменять иммунный тонус и метаболизм хозяина, имеют было сообщено. Например, при ожирении или вскоре после HFD проницаемость кишечника увеличивается, что делает возможной бактериальную транслокацию. через кишечный барьер (Neal et al. 2006; Amar et al. 2011; Ha et al. 2020). Более того, повышенная проницаемость кишечника также позволяет молекулам кишечной микробиоты, таким как липополисахарид (ЛПС), проникать. утечка в кровоток (Erridge et al.2007; Kumar et al. 2011). Повышенный LPS-стимулированный TLR4 вызывает провоспалительные реакции в различных иммуноцитах, эпителиальных клетках кишечника и инсулине. клетки-мишени, что приводит к инсулинорезистентности и непереносимости глюкозы (Shi et al. 2006; Saberi et al. 2009; Guo et al. 2013; Tao et al. 2017). Уровни в плазме другой молекулы, производной от кишечной микробиоты, формилметиониллейцилфенилаланина (формилпептиды N- ), также повышены у мышей с HFD / ожирением. Генетическая делеция или фармакологическое ингибирование рецептора формилпептида N- Fpr1 улучшает толерантность к глюкозе за счет увеличения глюкагоноподобного пептида на 1 участок от энтероэндокрина L-клетки с последующим увеличением GSIS β-клеток.Из биоактивных метаболитов, продуцируемых микробиотой кишечника, короткоцепочечные жирные кислоты (SCFAs) являются одними из самых распространенных (Stevens and Hume 1998). SCFAs активируют рецепторы, связанные с G-белком (GPCR), включая GPR41 и GPR43, которые экспрессируются в адипоцитах, энтероцитах, иммуноциты и β-клетки поджелудочной железы. В нескольких отчетах показано положительное влияние активации GPR43 на снижение ожирения и улучшают чувствительность к инсулину, а также GSIS β-клеток у мышей с ожирением (Tolhurst et al.2012; Kimura et al. 2013; McNelis et al. 2015). Однако также сообщалось о противоречивых результатах (для обзора см. Ang and Ding 2016): например, β-клеточное истощение как GPR41, так и GPR43 увеличивает GSIS и улучшает толерантность к глюкозе у мышей (Tang et al. 2015). Помимо НЖК, вторичные и третичные желчные кислоты, продуцируемые кишечными микробами, изменяют состав желчной кислоты. бассейн (Brufau et al. 2010; Suhre et al. 2010; Haeusler et al. 2013; Ridaura et al. 2013; Yoshimoto et al.2013) и модулируют воспаление и метаболизм, стимулируя рецептор желчной кислоты 1 (TGR5) и рецептор фарнезоида X (FXR) (Pols et al. 2011; Wollam and Antebi 2011).
Вместе взятые, желудочно-кишечный тракт играет роль в связанном с ожирением хроническом воспалении тканей и нарушении регуляции метаболизма. Один очевидным механизмом является то, что развитие ожирения приводит к повышенной проницаемости желудочно-кишечного тракта, так что провоспалительные бактериальные продукты (например,g., LPS), и даже сами бактерии получают доступ к окружающим лимфатическим узлам и системному кровообращению. Кроме того, дисбактериоз ожирения обеспечивает еще один потенциальный механизм, при котором определенные виды бактерий ЖКТ или их комбинации видов, может способствовать мета воспламенению. Последний виден у мышей, но потребует дополнительной проверки трансляции. в людях. Это многообещающая область, поскольку дальнейшие исследования могут выявить конкретные факторы, которые могут быть полезны при открытии лекарств.
Гипоксия как исходное событие
Как отмечалось выше, хорошо известно, что ожирение приводит к накоплению провоспалительных макрофагов в жировой ткани в как мыши, так и люди. Эти банкоматы являются ключевыми факторами развития инсулинорезистентности и непереносимости глюкозы. Области В настоящее время активный интерес сосредоточен на том, как начинается накопление макрофагов в начале ожирения и как действуют провоспалительные Банкоматы вызывают системную метаболическую дисфункцию.Было опубликовано несколько исследований, дающих важную информацию по этим вопросам. В этом разделе мы попытаемся объединить эти новые идеи в общую последовательную гипотезу.
В течение многих лет было известно, что кислородное напряжение жировой ткани снижается при ожирении как у мышей, так и у людей (Hosogai et al. 2007; Halberg et al. 2009; Pasarica et al. 2009, 2010; Lawler et al. 2016; Seo et al.2019; Smith et al.2019). Первоначально предполагалось, что гипоксическое состояние жировой ткани связано с дисбалансом между увеличением массы жировой ткани. и доступная доставка кислорода.Однако напряжение кислорода в тканях поддерживается за счет баланса между поставкой и потреблением кислорода, и более свежие данные показывают, что ожирение приводит к увеличению потребления кислорода адипоцитами (Lee et al. 2014). Это повышенное потребление составляет около 40% снижения межклеточного давления кислорода при ожирении (Seo et al.2019). Остающийся компонент пониженного межклеточного давления кислорода не связан со снижением доставки кислорода в артериальную кровь (кровь поток в жировые отложения и насыщение крови O 2 ), но это объясняется снижением функциональной плотности капилляров в жировой ткани с ожирением (Lee et al.2014). Следовательно, есть два компонента: снижение плотности капилляров и повышенное потребление кислорода адипоцитами, которые способствуют к относительной гипоксии жировой ткани при ожирении. Как правило, гипоксические состояния приводят к большему выражению фактор транскрипции адипоцитов HIF-1α, запускающий последующий ответ транскриптома гипоксии. Однако индукция HIF-1α не происходит до тех пор, пока напряжение кислорода не упадет до ≤1% (Seo et al.2019). Следовательно, снижение напряжения кислорода во внеклеточной жировой ткани, описанное при ожирении, вероятно, недостаточно для запуска гипоксический ответ на HIF-1α. С другой стороны, увеличение потребления кислорода адипоцитами весьма заметно при ожирении, в результате чего концентрация кислорода внутри адипоцитов до уровней ниже ~ 1,4%. Этого достаточно для индукции экспрессии HIF-1α в адипоцитах. и последующие эффекты, опосредованные этим фактором транскрипции (Hosogai et al.2007; Halberg et al. 2009; Ли и др. 2014). Эта точка зрения подтверждается открытием, что индуцированные гипоксией аддукты пимонидазола в ожирении жировой ткани ограничиваются адипоциты (Lee et al., 2014). Взятые вместе, увеличение потребления кислорода адипоцитами, вызванное ожирением, вероятно, является основным фактором внутриклеточного гипоксический ответ адипоцитов, в то время как снижение интерстициального давления кислорода, вероятно, играет вспомогательную или разрешающую роль в преувеличении влияния повышенного потребления кислорода адипоцитами.
Механизмы, с помощью которых уровни внутриклеточного кислорода регулируют HIF-1α, хорошо описаны и происходят в основном в белке уровень с меньшим влиянием на экспрессию мРНК (Иван и Келин, 2017; Шедель и Ратклифф, 2019; Семенза, 2019). В нормоксических условиях белки домена пролилгидроксилазы (PHD) связываются с HIF-1α, и PHD-зависимое гидроксилирование HIF-1α приводит к убиквитин-зависимой протеасомной деградации белка.В условиях гипоксии ПГД инактивируются, что приводит к Стабилизация белка HIF-1α, вызывающая повышенную экспрессию белка HIF-1α. Хотя это основная движущая сила регулирования экспрессии HIF-1α, при ожирении, умеренное увеличение мРНК HIF-1α также сообщалось как способствующий фактор (Lee et al. 2014).
Эта индукция HIF-1α адипоцитов играет важную роль в опосредовании общего воспаления жировой ткани (Lee et al.2014). Это может происходить с помощью нескольких механизмов. Например, несколько хемокинов являются нижестоящими мишенями транскрипции HIF-1α, и экспрессия этих хемокинов увеличивается при ожирении и других состояниях гипоксии. Эти хемокины служат для набора моноциты в гипоксическую жировую ткань, которые затем преимущественно дифференцируются в провоспалительные, M1-подобные ATM. Как обсуждалось более подробно выше, увеличение количества M1-подобных ATM является ключевым механизмом метаболической дисфункции, характеризуемой инсулином. резистентность и непереносимость глюкозы при ожирении (рис.3). Есть много исследований, в которых различные провоспалительные гены были удалены в макрофагах, и все эти исследования постоянно демонстрируют улучшение метаболической дисфункции, вызванной ожирением (Arkan et al. 2005; Weisberg et al. 2006; Saberi et al. 2009; Han et al. 2013; Li et al. 2015). В соответствии с этой формулировкой центральная роль HIF-1α в провоспалительных ответах, ведущих к инсулинорезистентности было показано в исследованиях нокаута по типу клеток (Jiang et al.2011; Ли и др. 2011а, 2014; Кришнан и др. 2012; Sun et al. 2013). Таким образом, специфический для адипоцитов нокаут HIF-1α в значительной степени предотвращает индуцированное ожирением накопление АТМ, воспаление жировой ткани, непереносимость глюкозы и инсулинорезистентность, но не вызывает никаких изменений в общей массе тела. Это подчеркивает важность HIF-1α в общем воспалительном ответе жировой ткани, что согласуется с идеей о том, что внутриклеточный гипоксия адипоцитов приводит к индукции HIF-1α, что, в свою очередь, способствует накоплению АТМ, воспалению и последующему развитию глюкозы. непереносимость и инсулинорезистентность.
Рисунок 3.Внутриклеточная гипоксия адипоцитов вызывает воспаление и инсулинорезистентность. При ожирении повышенное содержание внутриклеточных СЖК стимулирует ANT2-зависимое усиление митохондриального несвязанного дыхания в адипоцитах. В сочетании с пониженной функциональной плотностью капилляров, это приводит к внутриклеточной гипоксии и стабилизации HIF-1α.HIF-1α индуцирует повышенную продукцию хемокинов, что приводит к увеличению инфильтрация иммунных клеток, включая моноциты, и повышенная пролиферация ATM. Эти изменения критически влияют на экзосомы банкомата. секреция, а также производство экзосом адипоцитов и адипокинов, вызывающих системную инсулинорезистентность. Большинство из этих последовательных события были показаны в моделях мыши. Эта гипотеза еще предстоит подтвердить на людях.
Также представляют интерес механизмы, лежащие в основе повышенного потребления кислорода адипоцитами, вызванного ожирением.Адениновый нуклеотид транслоказа (ANT) 2 — это белок внутренней мембраны митохондрий, который перекачивает протоны из межмембранного пространства в митохондриальную матрица (Бертолет и др., 2019). Следовательно, увеличение активности ANT2 приводит к утечке протонов из межмембранного пространства, что приводит к несвязанной митохондриальной дыхание с повышенным потреблением O 2 . Следует отметить, что другой важный разобщающий белок, UCP1, не экспрессируется в белых адипоцитах (Cousin et al.1992; Wu et al. 2012а). Уровни НЖК в жировой ткани повышаются при ожирении, и НЖК могут активировать ANT2-зависимое разобщение окислительного метаболизма, приводит к увеличению потребления кислорода адипоцитами (Lee et al. 2014). Теоретически повышенное окисление жирных кислот может также привести к большему потреблению кислорода, но более подробные исследования показали, что показали, что этот компонент весьма незначителен по сравнению с прямым действием НЖК на активацию белка ANT2. Для дальнейшего укрепить эту общую концепцию, удаление ANT2 в адипоцитах может обратить все эти гипоксические реакции.Таким образом, специфичные для адипоцитов ANT2 KO достаточно для блокирования вызванного ожирением увеличения потребления адипоцитов O 2 , улучшения реакции гипоксии адипоцитов и предотвращения индукции HIF-1α (Seo et al., 2019). Это смягчает развитие воспаления жировой ткани с пониженным содержанием провоспалительных АТМ и сниженной экспрессией. генов воспалительного пути. Это сопровождается значительным улучшением толерантности к глюкозе и чувствительности к инсулину у ожирение мышей без изменения массы тела.
Как воспаление жировой ткани вызывает инсулинорезистентность?
Тот факт, что повышенное содержание провоспалительных АТМ, вызванное ожирением, может напрямую вызывать инсулинорезистентность, хорошо известен. по крайней мере, у мышей, но не совсем ясно, как именно M1-подобные макрофаги приводят к снижению чувствительности к инсулину. был предметом интенсивного расследования.Логическое предположение состоит в том, что провоспалительные макрофаги выделяют факторы, которые может вызывать паракринные или системные эффекты на клетки-мишени инсулина, нарушая передачу сигналов инсулина. Классически M1-подобная поляризованная макрофаги выделяют множество цитокинов и хемокинов. Хемокины действуют, обеспечивая градиент концентрации, в котором моноциты и другие типы иммунных клеток могут мигрировать вниз по этому градиенту к источнику хемокина; это рабочее определение хемотаксиса.При ожирении некоторые из этих хемокинов, вырабатываемых жировой тканью, могут попадать в кровоток. Тем не мение, циркулирующие хемокины не способны создавать дифференциальный градиент концентрации, способствующий хемотаксису моноцитов. кровообращения в воспаленные ткани. Кроме того, циркулирующие уровни хемокинов из-за этого «побочного эффекта» обычно невысоки. намного ниже, чем биологически эффективные концентрации, поэтому они не будут иметь значимых системных физиологических эффектов.Даже если конкретный хемокин может вызывать снижение передачи сигналов инсулина in vitro (Sartipy and Loskutoff, 2003), все равно будет сложно сделать вывод о том, что такой хемокин может действовать системно, поскольку циркулирующий системный концентрации настолько низкие.
Больше внимания было уделено секретируемым цитокинам, так как хорошо известно, что некоторые цитокины (например, TNFα) обладают сильным действием. эффекты, чтобы вызвать снижение передачи сигналов инсулина.TNFα может вызывать местные тканевые эффекты, снижающие действие инсулина (Hotamisligil et al. 1994) за счет нескольких механизмов. TNFα может снижать экспрессию Irs2 и Glut4 , способствовать ингибирующему фосфорилированию белков субстрата инсулинового рецептора (IRS), усиливать липолиз адипоцитов. (Высвобождение FFA) и синтез церамидов, а также подавляют экспрессию PPARγ (Stephens et al. 1997; Guilherme et al. 2008). Это явление может происходить в различных отложениях жировой ткани, которые во время курса увеличиваются и воспаляются. ожирения (TNFα используется в качестве примера, поскольку это наиболее хорошо изученный цитокин, полученный из иммунных клеток).Однако системный инсулинорезистентность требует снижения чувствительности к инсулину в мышцах и печени, а также жировой ткани, и если только мышцы и печень вырабатывают высокие уровни тканевых цитокинов, маловероятно, что TNFα или другие цитокины, которые выходят из ожирение жировой ткани может вызвать системные эффекты. Таким образом, циркулирующие концентрации TNFα и других цитокинов существенно ниже. ниже уровней, необходимых для оказания биологических эффектов в тканях (Stephens et al.1997; Amar et al. 2007; McGillicuddy et al. 2011). Напротив, уровни цитокинов в жировой ткани могут быть значительно повышены при ожирении, и это может вызвать местные эффекты. Если вводят большие количества TNFα in vivo, повышая уровни циркуляции намного выше, чем они обычно существуют при ожирении, затем наступает инсулинорезистентное состояние (Lang et al. 1992). Однако это не отражает фактическое патофизиологическое состояние ожирения, при котором уровни цитокинов в крови, а выше, чем у нормальных людей, не повышаются до уровней, при которых они оказывают существенное биологическое воздействие.Ил-6 может быть исключением этого общего принципа, поскольку уровни IL-6, которые попадают в кровоток при ожирении, как сообщается, биологически активный. Однако исследования метаболической активности IL-6 смешиваются с некоторыми исследованиями, показывающими, что инсулиноподобное действие улучшает чувствительность к инсулину, в то время как другие сообщают, что она подавляет действие инсулина (Carey et al. 2006; Franckhauser et al. 2008).
Учитывая приведенное выше обсуждение, многие лаборатории пытались определить другие факторы, разработанные на основе M1-подобных макрофаги, которые могут вызвать метаболическую дисфункцию.Одним из таких факторов является Галектин-3. Галектин-3 производится почти исключительно от макрофагов, а также при ожирении как у людей, так и у мышей, уровни в крови довольно высоки по сравнению с нормальными и находятся в диапазоне где они оказывают биологическое воздействие (Li et al., 2016). Действительно, у людей циркулирующие уровни галектина-3 повышены по сравнению с нормальными субъектами, и степень повышения Уровень галектина-3 коррелирует с величиной ожирения и степенью инсулинорезистентности.Исследования in vitro показывают что обработка галектином-3 клеток-мишеней инсулина может напрямую вызывать снижение передачи сигналов инсулина, нарушая способность инсулина, чтобы правильно активировать рецептор инсулина на поверхности клетки. Исследования потери функции были выполнены анализ макрофаг-специфичных мышей Galectin-3 KO. Истощение макрофагов галектина-3 защищает мышей HFD от развития непереносимости глюкозы и инсулинорезистентности, тогда как прибавка в весе вполне нормальна.Дополнительное усиление и потеря функции исследования с галектином-3 согласуются с этой формулировкой, предполагая, что циркулирующий галектин-3, происходящий из макрофагов, может способствуют развитию инсулинорезистентности при ожирении.
Приведенные выше соображения привели к поиску дополнительных факторов, которые могут попасть в кровоток из ожирения жировой ткани. и вызывают системную резистентность к инсулину.В самом деле, недавние исследования показывают, что экзосомы, происходящие из ATM, могут выполнять эту роль. Экзосомы представляют собой небольшие наночастицы (∼150 нм), продуцируемые большинством типов клеток (Pegtel and Gould, 2019). Они могут секретироваться в интерстициальное пространство, где затем попадают в кровоток, приводя как к паракринным, так и к эндокринным типам. последствия. Экзосомы содержат множество компонентов груза, включая белки, липиды, микроРНК (миРНК) и мРНК, а также как ряд других видов РНК.Недавние исследования показали, что экзосомы, полученные из банкоматов у тучных мышей, могут напрямую вызывать инсулинорезистентность in vitro при применении к адипоцитам, первичным гепатоцитам или миоцитам (Ying et al. 2017a). Эти «тучные» экзосомы, полученные из ATM, можно собирать и вводить внутривенно худым мышам. Когда это было сделано, У худых мышей-реципиентов развилась непереносимость глюкозы, гиперинсулинемия и инсулинорезистентность, сравнимые с таковыми у мышей-реципиентов. ожирение.Важно отметить, что пищевое поведение и масса тела не меняются в результате обработки экзосомами in vivo. С другой Сторона этой медали, экзосомы ATM, собранные у тощих мышей, получавших корм, дают противоположный фенотип. Лечение адипоцитов, миоциты или первичные гепатоциты с «худыми» экзосомами ATM напрямую приводят к усилению передачи сигналов инсулина. Важнее, когда тучных мышей лечили «худыми» экзосомами, полученными из АТМ, в течение 3 недель, у этих мышей было заметно улучшенное содержание глюкозы. переносимость при повышении чувствительности к инсулину.В самом прямом смысле эти «тощие» экзосомы ATM вызывают терапевтический ответ, смягчение метаболической дисфункции при ожирении. Что касается механизмов, эти исследования также показали, что биологические эффекты экзосом ATM с ожирением и худыми полностью опосредуются их грузом miRNA, поскольку истощение экзосомальных miRNA предотвращает все этих действий, опосредованных экзосомами (рис. 4).
Рисунок 4.Системное действие экзосом АТМ. Банкоматы могут регулировать системную чувствительность к инсулину, высвобождая miRNA-содержащие экзосомы. Инъекция экзосом, высвобождаемых из банкоматов худых мышей, увеличивает чувствительность к инсулину у тучных мышей, тогда как инъекция тучных мышей, полученных из банкоматов экзосомы вызывают инсулинорезистентность у худых мышей. Кроме того, прямая обработка инсулиновых клеток-мишеней in vitro с помощью «постного» или «тучные» экзосомы напрямую вызывают чувствительность или резистентность к инсулину, соответственно.Большинство этих последовательных событий имеют были показаны в моделях мышей. Эта гипотеза еще предстоит подтвердить на людях.
Недавние исследования уточнили конкретные miRNA, которые могут вызывать эти полезные «терапевтические» эффекты у мышей. Проводя полное секвенирование экзосом, полученных из M2-подобных ATM, в сочетании с биоинформатическим подходом и скринингом конкретных мРНК miR-690, активированная в худых экзосомах, происходящих из ATM, была идентифицирована как miRNA, сенсибилизирующая инсулин.Таким образом, делеция miR-690 предотвращает благоприятные эффекты экзосом M2-подобных макрофагов, а искусственный миметик miR-690 воспроизводит полезные влияние худых экзосом ATM на чувствительность к инсулину и непереносимость глюкозы. Кроме того, ингибитор (антагомир) miR-690 полностью блокирует эффекты миметика miR-690. Таким образом, miR-690 выступает как настоящая инсулино-сенсибилизирующая молекула в обоих клетки мыши и человека, и если трансляционные исследования будут успешными, эта молекула потенциально может привести к сенсибилизации к инсулину. терапевтический.
Комплексное исследование Thomou et al. (2017) идентифицировали присутствие нескольких сотен miRNA в экзосомах крови мышей WT. Когда они изучали адипоцит-специфический Dicer KO-мышей, чтобы истощить все миРНК из любых происходящих из адипоцитов экзосом, которые попадают в кровоток, они обнаружили, что ~ 60% циркулирующие миРНК были уменьшены, что указывает на то, что адипоциты вносят важный количественный вклад в циркулирующие экзосомы. бассейн.Это исследование также показало, что miR-99b высоко экспрессируется в экзосомах, высвобождаемых из коричневых адипоцитов, и что miR-99b может подавляют экспрессию FGF21 в печени и предположили, что снижение уровней циркулирующего FGF21 связано с улучшением толерантность к глюкозе они наблюдали у мышей Dicer KO с адипоцитами. Основываясь на исследованиях секвенирования miRNA, важно отметить: что существует заметная разница в составе миРНК между АТМ и экзосомами, полученными из адипоцитов, с перекрыванием только на ~ 20%. согласованность (Thomou et al.2017; Ying et al. 2017а). Это дополнительно подчеркивает необходимость понимания функции экзосом, специфичных для определенного типа клеток, при метаболических заболеваниях.
Экзосомы являются компонентом более разнообразной группы секретируемых частиц, называемых внеклеточными везикулами (EV). Есть несколько дополнительные опубликованные исследования, показывающие результаты, согласующиеся с точкой зрения, что ЭВ / экзосомы, полученные из ожирения жировой ткани может влиять на чувствительность к инсулину (Kita et al.2019). Например, препараты EV / экзосомы жировой ткани человека с ожирением демонстрировали дифференциальную экспрессию нескольких miRNA, которые может повлиять на передачу сигналов инсулина. Экзосомы, полученные непосредственно из жировой ткани ожирения, ухудшают чувствительность к инсулину, и одно исследование предполагают, что miR141-3P играет причинную роль (Kranendonk et al. 2014; Dang et al. 2019). Следует отметить, что в исследованиях с использованием экзосом, полученных из жировой ткани, неизвестно, какой тип клеток в жировой ткани ткань производит биологически активные частицы.
Взятые вместе, вышеупомянутые исследования приводят к модели, изображенной на Рисунке 3. Эта модель демонстрирует общую последовательную гипотезу, в которой ожирение приводит к повышенной активации НЖК ANT2, вызывая несвязанные дыхание с повышенным потреблением кислорода адипоцитами. Это приводит к состоянию относительной гипоксии внутри адипоцита, вызывая Индукция HIF-1α и последующие воспалительные реакции, включая набор и накопление ATM и повышенную экспрессию множества провоспалительных генов.Это воспалительное состояние затем может привести к возникновению непереносимости инсулина и глюкозы, и происходящие из ATM экзосомальные miRNA (особенно miR-690) являются, по крайней мере, одним механизмом, приводящим к метаболической дисфункции, которая характеризует ожирение.
Клинические последствия
В то время как различные эксперименты с генетическим набором и потерей функции на мышах ясно показали важность хронического воспаления тканей. Что касается этиологии инсулинорезистентности и непереносимости глюкозы, концепция еще требует окончательного подтверждения на людях.Поскольку такие виды исследований манипуляции генами невозможны на людях, нам нужно будет полагаться на различные противовоспалительные средства. методы фармакологического лечения, чтобы окончательно проверить эту общую концепцию на людях.
По мере того, как мы уточняем наши механистические представления о последствиях хронического воспаления, мы получаем более точные инструменты для проверки всего этого. Мы надеемся, что концепции у людей появятся. Хотя это еще предстоит сделать, в настоящее время проводится ряд клинических исследований. предоставление данных, относящихся к этой проблеме.Например, есть несколько сообщений об использовании ингибирующих антител, направленных против TNFα у пациентов с нарушением обмена веществ. Очевидно, что эти агенты имеют большой клинический успех при лечении ревматоидных заболеваний. артрит и воспалительное заболевание кишечника (Lin et al., 2020; Vulliemoz et al., 2020), и многие пациенты, получающие лечение от этих других состояний, страдают ожирением или диабетом 2 типа. Некоторые из этих пациентов были проанализированы, сообщалось о небольших клинических исследованиях.К сожалению, результаты этих исследований были разными. появляются однозначные выводы. В некоторых отчетах отмечается умеренное улучшение гликемии или чувствительности к инсулину (Kiortsis et al. 2005; Gonzalez-Gay et al. 2006), тогда как в других не обнаружено никаких эффектов (Ofei et al. 1996; Paquot et al. 2000; Dominguez et al. 2005; Бернштейн и др., 2006; Вашер и др., 2011). Потребуются крупномасштабные рандомизированные двойные слепые клинические испытания, чтобы выявить любые возможные эффекты ингибирования TNFα. при нарушении обмена веществ.Возможно, наиболее многообещающие результаты получены из метаанализа, показывающего, что лечение антителами TNF-α может снизить риск развития T2DM (Burska et al. 2015), хотя, опять же, это должно быть продемонстрировано в проспективном исследовании для трансляционной валидации. На основе имеющихся данных, разумно предположить, что эффекты лечения анти-TNFα могут не быть клинически устойчивыми при метаболических заболеваниях, с рядом исследований, показывающих маргинальные эффекты или их отсутствие.При отсутствии крупных проспективных исследований полезно рассмотреть почему ингибирование TNFα может быть не очень эффективным у людей, как это было у мышей с ожирением. Одна из возможностей заключается в том, что у людей TNFα не так важен для инсулинорезистентности, как у мышей, и это еще предстоит проверить. Другой сценарий заключается в том, что цитокины, такие как TNFα, работают преимущественно на тканевом уровне, а не через эффекты циркулирующего TNFα, и это возможно, что биопрепараты против TNFα не проникают в жировую ткань при достаточно высоких концентрациях, чтобы в достаточной степени блокировать высокие уровни TNFα в тканях при ожирении.
Сообщалось также об ингибирующих антителах, направленных против другого воспалительного цитокина, IL-1β. Некоторые из этих исследований были более масштабными и перспективными и показали небольшое, но последовательное снижение уровней глюкозы и HbA1c в результате ингибирования IL-1β (Ларсен и др., 2007; Эверетт и др., 2018). Однако эти положительные эффекты, по-видимому, связаны с усилением секреции инсулина и функцией β-клеток, а не с улучшением в чувствительности к инсулину.Это может указывать на то, что IL-1β играет значительную роль в развитии дисфункции β-клеток. при СД2, но механистически не имеет отношения к возникновению инсулинорезистентности, вызванной воспалением. Масштабная перспектива Сообщалось о клиническом исследовании лечения сердечно-сосудистых заболеваний, названном испытанием Cantos, которое показало клинически значимое снижение частоты сердечно-сосудистых событий у пациентов, получавших антитела к IL-1β (Ridker et al.2017). Хотя имелись некоторые серьезные нежелательные побочные эффекты, связанные с инфекциями, это исследование действительно подчеркивает потенциальные полезность противовоспалительной терапии при атеросклерозе.
Другой потенциальный противовоспалительный механизм с использованием производных салициловой кислоты также изучался в контексте инсулина. резистентность и СД2. Ранние мелкомасштабные исследования показали многообещающие эффекты лечения салсалатом для улучшения чувствительности к инсулину. и снизить гликемию при СД2 (Goldfine et al.2008 г.). Последующее крупномасштабное проспективное исследование также показало умеренное снижение уровня HbA1c на 0,3–0,5% при диабете. (Голдфайн и др., 2010). Хотя это обнадеживает, за последние несколько лет в клинических исследованиях салицилового кислотная производная обработка. Кроме того, следует отметить, что, хотя изначально считалось, что салицилаты обладают противовоспалительным действием, путем ингибирования IKKβ (Yuan et al.2001), другие исследования поставили под сомнение, является ли это механизм действия этого класса препаратов. Таким образом, некоторые отчеты показали что салицилаты могут активировать AMPK, что приводит к улучшению метаболизма (Hawley et al. 2012).
Были предложены другие возможные подходы. К ним относятся разработка низкомолекулярного агониста GPR120 для использования противовоспалительные, инсулино-сенсибилизирующие эффекты этого сигнального каскада.Возможно ингибиторы циркулирующего галектина-3 или селективное ингибирование хемотаксиса моноцитов в жировую ткань и / или печень также может оказаться полезным. Хотя возможно в будущем пока не появилось никаких программ клинических разработок, связанных с этими механизмами.
Поскольку воспаление печени является важным компонентом патофизиологии НАСГ, полезно кратко описать состояние специфических противовоспалительных методов лечения этого заболевания.При НАСГ были протестированы два противовоспалительных механизма: Ингибирование ASK1 (Харрисон и др., 2020) и ингибирование CCR2 / 5 (Ратциу и др., 2020). Хотя долгосрочные клинические исследования фазы 3 не показали какого-либо улучшения показателей фиброза или НАСГ, это важно. чтобы указать, что, хотя эти методы лечения могут быть «потенциальными противовоспалительными», на самом деле они не вызывают каких-либо противовоспалительных эффектов. эффекты в клинических испытаниях. Другими словами, баллы НАСГ, которые включают лобулярное воспаление, не изменились в Пациенты, получавшие ингибиторы ASK1 и CCR2 / 5.При оценке НАСГ воспаление оценивается отдельно по шкале от 1 до 4 и средней значение в этих исследованиях было ~ 2. Эффективная терапия должна снизить оценку воспаления по крайней мере на один балл у леченных субъектов. и в этих отчетах этого не было. Следовательно, как и в случае неэффективности «противовоспалительного» лечения инсулинорезистентности, нужно понимать, что просто называть лечение противовоспалительным и не обнаруживать эффекта на НАСГ или инсулинорезистентность. не означает, что лечение было действительно противовоспалительным.Опубликованные клинические данные просто не обеспечивают эффективного противовоспалительный механизм для проверки гипотезы. Другими словами, отсутствие положительных данных — это не то же самое, что наличие отрицательных данных.
С точки зрения трансляции, важно помнить, что подавление воспалительных реакций может привести к подавлению специфических аспектов иммунной системы, которые могут привести к нежелательным последствиям для безопасности.За этим нужно внимательно следить в любой программе клинической разработки, включающей противовоспалительный подход к лечению метаболических заболеваний. Это также информирует виды стратегий, используемых для определения мишеней для лекарств или лекарств, которые вызывают сенсибилизацию к инсулину и противовоспалительное действие. Проксимальные провоспалительные сигналы обычно активируют ряд перекрывающихся и взаимосвязанных воспалительных путей, повышая возможность того, что нацеливание на конкретное дистальное воспалительное событие может иметь неадекватную эффективность, если избыточное провоспалительное действие сигнальные системы остались нетронутыми.С другой стороны, подавление более широкой сети воспалительных путей может вероятно несут риск увеличения нежелательных побочных эффектов. В этом контексте было бы выгоднее направить терапию в конкретном аспекте воспалительной реакции, критической для развития инсулинорезистентности. Очевидно, если препарат был разработан способ доставки, обеспечивающий тканевую селективность, это было бы большим преимуществом в предотвращении инсулинорезистентности. с меньшим риском нежелательных побочных эффектов.
Тиазолидиндионы (TZD) представляют собой высокоэффективный класс антидиабетических препаратов, которые стимулируют PPARγ и повышают уровень инсулина. сопротивление. TZD обладают противовоспалительным действием, напрямую ингибируя провоспалительную активацию макрофагов, способствуя дифференцировка в сторону противовоспалительного M2-подобного состояния (Jiang et al. 1998; Ricote et al. 1998; Bouhlel et al. 2007; Nelson et al. 2018). Кроме того, TZD могут действовать на PPARγ внутри Treg, стимулируя эти клетки ограничивать воспаление, вызванное врожденными иммунная система (Cipolletta et al.2012). С другой стороны, TZD изучаются в течение многих лет, и ряд дополнительных механизмов TZD / PPARγ-индуцированного инсулина чувствительность не выявлена (Tontonoz and Spiegelman 2008). К ним относятся стимуляция высвобождения адипонектина (Yu et al., 2002; Xia et al., 2018), перераспределение запасов жира в жировой ткани (de Souza et al., 2001) и индукция прямых генов PPARγ (таких как Glut-4) (Young et al. 1995), которые опосредуют передачу сигналов инсулина. В клинической ситуации in vivo в настоящее время невозможно определить степень противовоспалительные эффекты, опосредованные TZD, способствуют общему сенсибилизирующему к инсулину, антидиабетическому действию этих агенты.
Поскольку ожирение является ключевым патофизиологическим фактором в развитии хронического воспаления тканей, инсулинорезистентности и в конечном итоге СД2 и / или НАСГ, очевидно, что устойчивая потеря веса является прямым методом профилактики или лечения для этих условий. К сожалению, хотя этот метод лечения может быть простым, он далеко не из легких. Есть бариатрические хирургические подходы, несколько медикаментозных методов лечения ожирения и многочисленные формы изменения образа жизни, сочетающие различные диеты с упражнениями.К сожалению, эффекты потери веса часто бывают недолговечными, и восстановление веса (рецидив) является непростой задачей. основная нерешенная проблема с текущими фармакологическими подходами и подходами к образу жизни. Даже потеря веса после бариатрической операции может со временем ослабевают, хотя, в целом, снижение веса, достигаемое с помощью хирургических подходов, намного более длительное, чем с помощью образ жизни и лечение наркозависимости. Несмотря на вышеупомянутые трудности, связанные с длительным похудением, его в избытке ясно, что снижение веса приводит к большим терапевтическим эффектам в отношении уменьшения воспаления тканей, улучшения СД2, а также профилактика или лечение НАЖБП / НАСГ.Поскольку потеря веса приводит к этим множественным положительным эффектам одновременно, невозможно выделить вклад воспаления тканей в общее улучшение метаболизма, вызванное потеря веса, так как при любом уменьшении ожирения меняются несколько факторов.
Как обсуждалось ранее, наличие повышенных провоспалительных банкоматов, а также других иммунных клеток было хорошо задокументировано. при ожирении у человека, а также при наличии повышенной воспалительной реакции в печени, островках поджелудочной железы и желудочно-кишечном тракте. тракт.На данный момент результаты трансляции представляют собой ассоциации и корреляции и не доказывают причинно-следственную связь. Поскольку генетический манипуляции с людьми невозможны, потребуется эффективное противовоспалительное средство для проверки этой общей концепции. Как резюмировано выше, было опробовано небольшое количество потенциальных противовоспалительных подходов с ограниченным успехом. Следовательно, в этой области ожидаются дальнейшие специфические противовоспалительные методы лечения, чтобы лучше понять роль хронического воспаления тканей. в этиологии нарушения метаболизма у людей с ожирением.
Отчет о конкурирующих процентах
Авторы заявляют об отсутствии конкурирующих интересов.
Благодарности
Это исследование было поддержано Национальным институтом диабета, болезней органов пищеварения и почек Национальных институтов. здравоохранения (R01DK033651, P01DK074868, P30DK063491 и DK09062 на J.М.О. и R01DK124298 для Y.S.L.) и грант от Janssen Pharmaceuticals, Inc.
Нейтрофильная инфильтрация в модели воспаления ткани «орган на чипе»
Многофазная этиология воспаления тканей и фундаментальные иммунологические различия между видами затрудняют повторение воспалительных патологий на животных моделях и объясняют нехватку методов лечения, которые успешно переносятся с грызунов на человека.Здесь мы представляем подходящую для человека платформу «орган на чипе» для экспериментальных воспалительных заболеваний. Мы создали иммунокомпетентную модель кишечника in vitro путем включения эпителиальных и иммунных клеток кишечника в микрофлюидные камеры, которые позволяют клеткам перемещаться через внеклеточный матрикс (ЕСМ) и жидкостные каналы. Это первая модель, которая объединяет слизистый барьер, трехмерный ECM, резидентные и инфильтрирующие иммунные клетки и моделирует функциональные перекрестные помехи, которые в конечном итоге запускают клеточные процессы, характерные для воспаления.В гомеостатических условиях энтероциты образуют плотный эпителий, а субэпителиальные макрофаги не активируются. Введение провоспалительных медиаторов вызывает активацию макрофагов и вызванную воспалением проницаемость кишечного барьера. Нейтрофилы в параллельном неэпителиальном канале, отделенном от матрикса, привлекаются таким провоспалительным микроокружением и мигрируют через внеклеточный матрикс, дополнительно усугубляя воспаление и повреждение тканей. С помощью этой модели мы обеспечиваем основу для резюмирования и исследования начала воспаления тканей в контролируемой, релевантной для человека системе.
У вас есть доступ к этой статье
Подождите, пока мы загрузим ваш контент… Что-то пошло не так. Попробуй снова? .