Причины развития, симптомы атрофии мышц, принципы и методы лечения
Симптомы и лечение атрофии мышц ног и рук
Атрофией мышц называют истончение их волокон и уменьшение общего мышечного объема. При этом мышечная ткань может заменяться соединительной тканью, которая не способна сокращаться. Именно так развивается атрофия мышц кисти, голени и других частей тела.
Причины развития атрофии мышц
Рассматриваемое патологическое состояние может развиться при воздействии различных провоцирующих факторов. Чаще всего атрофия мышц наблюдается у пожилых людей, что связано с естественными процессами старения: замедляется метаболизм (обмен веществ в мышечных тканях), снижается физическая нагрузка. Но есть и другие причины развития патологии:
- нарушение гормонального баланса организма – оно может развиться при патологиях поджелудочной и щитовидной железы, надпочечников и яичников;
- заболевания органов желудочно-кишечного тракта;
- патология соединительной ткани;
- поражения периферических нервов.

В некоторых случаях спровоцировать проблему могут наследственные заболевания – например, причиной спинальной атрофии мышц являются генетические изменения в структурах нервной системы, которые делают невозможными произвольные сокращения мышц. Такие заболевания проявляются проблемами с мышцами уже в раннем детстве. Нередко именно атрофия мышц является единственным выраженным признаком заболевания, что нужно учесть в диагностике.
Немаловажную роль в развитии атрофии мышц играют питание и образ жизни – недостаток витаминов и микроэлементов в организме, злоупотребление алкоголем со временем приводят к рассматриваемой патологии.
Симптомы атрофии мышц
Лечению атрофии мышц ног и рук должна предшествовать диагностика патологии. Врачи подчеркивают, что необходимо обращать внимание на такие признаки:
- быстро наступающая усталость;
- слабость в мышцах на фоне физической нагрузки;
- уменьшение объема мышц;
- трудности при выполнении физической работы.

Симптомы атрофии мышц никогда не появляются остро, они характеризуются постепенным нарастанием. Патология развивается постепенно, могут пройти годы, прежде чем она будет диагностирована. Но уменьшение объема пораженной мышцы может наблюдаться и на ранней стадии развития патологии.
Со временем атрофия мышц бедра приводит к невозможности долго и быстро ходить, подниматься по лестнице, может быть затруднен даже подъем с постели.
При атрофии сердечной мышцы пациент жалуется на неконтролируемое учащение или замедление сердцебиения, внезапные приливы жара к лицу, периодическое онемение пальцев верхних конечностей, одышку при физических нагрузках, а затем и в покое. Атрофические изменения в сердечной мышце считаются самыми опасными, так как при отсутствии адекватного лечения могут привести к летальному исходу.
Лечение атрофии мышц
Даже зная, какие заболевания сопровождаются атрофией мышц, врач должен назначить дополнительные методы обследования и только после постановки точного диагноза определит лечение.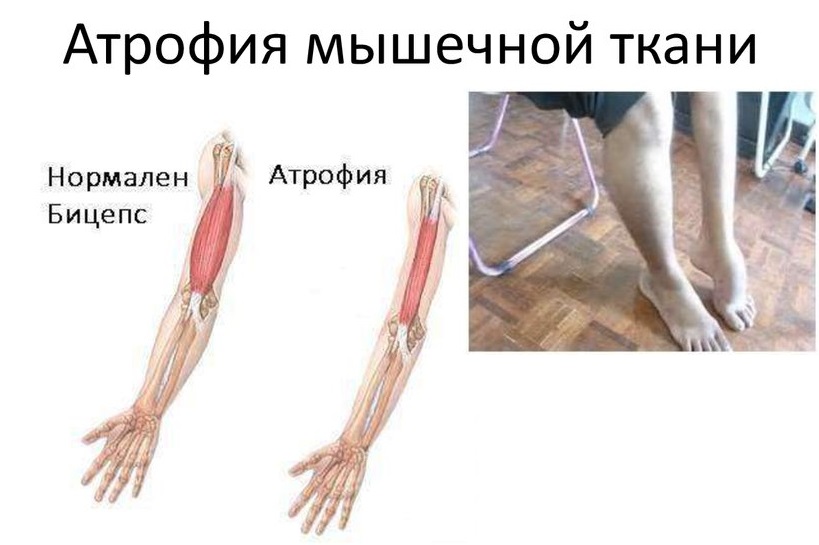 Часто после стабилизации общего состояния, остановки прогрессирования основной патологии либо переведения ее в состояние ремиссии исчезают и признаки атрофических изменений в мышечных тканях. При генетических заболеваниях полностью остановить прогрессирование описываемой патологии невозможно, но можно его замедлить.
Часто после стабилизации общего состояния, остановки прогрессирования основной патологии либо переведения ее в состояние ремиссии исчезают и признаки атрофических изменений в мышечных тканях. При генетических заболеваниях полностью остановить прогрессирование описываемой патологии невозможно, но можно его замедлить.
Общие принципы лечения атрофии мышц подразумевают проведение:
- медикаментозного лечения;
- физиотерапевтических процедур;
- занятия спортом.
Лекарственная терапия назначается в индивидуальном порядке. Это могут быть обезболивающие, нестероидные противовоспалительные средства, которые будут актуальными при сильном болевом синдроме. Индивидуальный подход особенно важен при выборе лечения патологии у детей – например, при лечении атрофии мышц голени у ребенка.
Важное значение в лечении имеют лечебная гимнастика и массаж. Упражнения для восстановления мышц при атрофии разрабатывают в индивидуальном порядке. Сначала они выполняются под контролем специалиста, дальнейшие занятия могут проводиться самостоятельно в домашних условиях.
Грамотное лечение, соблюдение всех рекомендаций врачей препятствуют прогрессированию патологии, больной может жить полноценной жизнью еще долгое время.
Более подробно о патологии, а также о том, кто из специалистов выполняет массаж при атрофии мышц нижних конечностей, можно узнать на страницах нашего сайта Добробут.ком.
Сестринское тест 2 — Стр 6
621. Центральный орган иммунной системы
а) пейеровы бляшки в тонком кишечнике
б) красный костный мозг
в) селезенка
г) периферические лимфоузлы
622. Периферический орган иммунной системы
а) селезенка
б) красный костный мозг
в) печень
г) вилочковая железа
623. Орган иммунной системы
а) щитовидная железа
б) вилочковая железа — тимус
в) поджелудочная железа
г) гипофиз
624. Причина гибели
Т4-лимфоцитов при ВИЧ-инфекции
Причина гибели
Т4-лимфоцитов при ВИЧ-инфекции
а) действие токсинов
б) размножение в них ВИЧ
в) соприкосновение их с ВИЧ
г) проникновение в них ВИЧ
625. Наиболее частое клиническое проявление острой лихорадочной фазы во II стадии ВИЧ-инфекции (по классификации В.В.Покровского) напоминает
а) грипп
б) инфекционный мононуклеоз
в) скарлатину
г) дифтерию
626. Такие признаки, как парезы, нарушения координации движения, кахексия, слепота, могут проявиться (по клинической классификации В.В. Покровского) в стадии ВИЧ-инфекции
а) I
б) IIВ
в) IIIВ
г) IIА
627. Термин «СПИД»
а) идентичен ВИЧ
б) обозначает терминальную стадию ВИЧ-инфекции
в) стадию вторичных проявлений
г) стадию первичных проявлений
628. СПИД-маркерным заболеванием является
СПИД-маркерным заболеванием является
а) лямблиоз
б) пневмоцистная пневмония
в) стафилококковая инфекция
г) амебиаз
629. Ношение маски медицинским работникам стационара для больных СПИДом необходимо для
а) предотвращения заражения их ВИЧ-инфекцией контактным путем
б) предотвращения заражения их ВИЧ-инфекцией через слюну
в) защиты больного от заражения микрофлорой персонала
г) защиты персонала от заражения ВИЧ воздушно-капельным путем
630. Запретить грудное вскармливание новорожденных детей матерям, которые инфицированы ВИЧ
а) следует
б) не следует, если соски не имеют трещин
в) не следует, если полость рта новорожденных без повреждений
г) не следует, если не прорезались зубы
631. Риск рождения ВИЧ-инфицированного ребенка от ВИЧ-инфицированной матери
а) 100%
б) до 70%
в) до 30%
г) не превышает 1%
632.
а) аденовирусную инфекцию
б) бактериальную дизентерию
в) генерализованную герпетическую инфекцию
г) паратифы
633. При развитии ВИЧ-инфекции для общего анализа крови характерен
а) лейкоцитоз
б) эозинофилия
в) появление атипичных мононуклеаров
г) уменьшение абсолютного количества лимфоцитов
634. Метод лабораторной диагностики с целью обнаружения ВИЧ
а) иммуноблотинг
б) ПЦР
в) ИФА
г) посев на
питательные среды
635. Характерная для ВИЧ-инфицированных больных пневмония
а) стафилококковая
б) пневмоцистная
в) микоплазменная
г) вирусная
636. ВИЧ-инфицированные
ВИЧ-инфицированные
а) отстраняются от всех видов прививок
б) не прививаются живыми вакцинами
в) прививаются в первую очередь в соответствии с календарем прививок
г) прививаются по эпидемиологическим показаниям
637. Причина геморрагического инсульта
а) ревматизм
б) сахарный диабет
в) шейный остеохондроз
г) гипертоническая болезнь
638. Менингеальный симптом — это
а) симптом Бабинского
б) ригидность мышц затылка
в) симптом «свисающей головы»
г) симптом Чураева
639. Сознание больного при геморрагическом инсульте
а) сохранено
б) утрачено на короткое время
в) утрачено на длительное время (кома)
г) изменено по типу сумеречного
640. При транспортировке
больного с геморрагией в мозг необходимо
При транспортировке
больного с геморрагией в мозг необходимо
а) убрать из-под головы подушку
б) приподнять ноги
в) часто менять положение головы и туловища
г) избегать изменений положения головы
641. Приступообразные боли в одной половине лица, иногда со слезотечением, выделением слизи из носа, слюнотечением, возникают при
а) неврите лицевого нерва
б) невралгии тройничного нерва
в) шейном остеохондрозе
г) опухоли головного мозга
642.Симптом «заячий глаз», сглаженность лобных и носогубной складок на пораженной стороне, перекос рта в здоровую сторону характерны для
а) опухоли головного мозга
б) энцефалита
в) неврита лицевого нерва
г) острого нарушения мозгового кровообращения
643. Для спастического паралича характерно
Для спастического паралича характерно
а) снижение сухожильных рефлексов
б) атрофия мышц
в) наличие патологических рефлексов
г) снижение мышечного тонуса
644. Для периферического (вялого) паралича характерно
а) повышение сухожильных рефлексов
б) наличие патологических рефлексов
в) атрофия мышц
г) повышение мышечного тонуса
645. Спастический гемипарез — это нарушение двигательной функции в
а) обеих ногах
б) одной руке
в) одной ноге
г) руке и ноге с одной стороны
646. Невралгия — это
а) воспаление нерва
б) повреждение нерва
в) боль по ходу нерва
г) атрофия нерва
647. Неврит — это
Неврит — это
а) воспаление нерва
б) разрыв нерва
в) боль по ходу нерва
г) атрофия нерва
648. Клинический симптом пояснично-крестцового радикулита
а) ригидность затылочных мышц
б) симптом Ласега
в) симптом Горнера
г) симптом Брудзинского
649. Доказательное исследование, позволяющее поставить диагноз менингита
а) увеличение СОЭ крови
б) лейкоцитоз крови
в) изменение ликвора
г) лимфопения крови
650. Основной симптом миастении
а) судороги
б) мышечная утомляемость
в) головная боль
г) тремор кистей
651. Для болезни Паркинсона характерно
Для болезни Паркинсона характерно
а) умственная деградация
б) нарушение координации
в) тремор кистей
г) параличи конечностей
652. При геморрагическом инсульте необходимо
а) придать больному положение с опущенным головным концом
б) положить на голову пузырь со льдом
в) повернуть больного на бок
г) придать больному полусидячее положение
653. Нарастающие, упорные головные боли распирающего характера и явления застоя на глазном дне характерны для
а) энцефалита
б) менингита
в) опухоли головного мозга
г) рассеянного склероза
654. Характерный признак невралгии тройничного нерва
а) приступообразные боли в одной половине лица
б) ригидность затылочных мышц
в) рвота
г) отсутствие складок на лбу при поднимании бровей
655. Постельный
режим при остром нарушении мозгового
кровообращения соблюдается в течение
Постельный
режим при остром нарушении мозгового
кровообращения соблюдается в течение
а) 14 дней
б) 10 дней
в) 21 дня
г) 30 дней
656. Односторонние поражения: открытый глаз, слезотечение из него, опущен угол рта, не поднимается бровь – это поражение
а) тройничного нерва
б) лицевого нерва
в) седалищного нерва
г) блуждающего нерва
657. Симптом, характерный для тяжелого сотрясения головного мозга
а) головная боль
б) шум в ушах
в) ретроградная амнезия
г) головокружение
658. Атрофия мышц — это основной симптом
а) спастического паралича
б) вялого паралича
в) миастении
г) болезни Паркинсона
659. Ощущение «треска»
в шее при поворотах головы характерно
для
Ощущение «треска»
в шее при поворотах головы характерно
для
а) менингита
б) остеохондроза
в) энцефалита
г) опухоли головного мозга
660. Патологические рефлексы характерны для
а) вялого паралича
б) спастического паралича
в) менингита
г) энцефалита
661. Повышенное стремление к деятельности характерно для
а) маниакальной фазы маниакально-депрессивного психоза
б) депрессивной фазы маниакально-депрессивного психоза
в) эпилепсии
г) кататонического синдрома
662. Афазия – это
а) нарушение речи
б) одна из форм мутизма
в) проявление кататонии
г) нарушение глотания
663. Психическая
ятрогения — это болезненное состояние,
возникающее в результате неправильного
Психическая
ятрогения — это болезненное состояние,
возникающее в результате неправильного
а) медикаментозного лечения психического заболевания
б) определения диагноза психического заболевания
в) поведения медицинского работника в отношении больного
г) ухода за больным с психическим заболеванием
664. Кардинальный признак неврастении
а) истерический припадок
б) раздражительная слабость
в) навязчивые страхи
г) сумеречное состояние
665. Для купирования маниакального возбуждения применяют
а) аминазин
б) димедрол
в) кофеин
г) церебролизин
666. Наиболее общий симптом шизофрении
Наиболее общий симптом шизофрении
а) отгороженность, отрыв от реальности, погружение в мир собственных переживаний
б) маниакальное возбуждение
в) отвлекаемость
г) амбулаторного автоматизма
667. Психогении — заболевания, возникающие под влиянием
а) тяжелых травм головного мозга
б) психической травмы
в) инфекций головного мозга
г) алкогольной интоксикации
668. Для депрессивного синдрома характерно
а) слабоумие
б) гипотимия
в) эйфория
г) раздражительность
669. Болезненная бесчувственность характерна для
а) старческого слабоумия
б) шизофрении
в) неврастении
г) эпилепсии
670. Особенностью
шизофрении у детей является наличие
Особенностью
шизофрении у детей является наличие
а) развернутого бреда
б) судорожных припадков
в) ночных страхов
г) полной потери ориентировки
671. Решающее значение в развитии неврозов имеет
а) нейроинфекция
б) черепно-мозговая травма
в) сосудистые заболевания головного мозга
г) психогенный фактор
672. Общий симптом инволюционных (предстарческих) психозов
а) зрительные галлюцинации
б) чувство тревоги
в) эмоциональная тупость
г) бред
673. Повышенное настроение, ускоренный темп мышления, повышенная деятельность характеризуют синдром
а) депрессивный
б) тревожно-депрессивный
в) маниакальный
г) судорожный
674. При ипохондрическом
бреде больной считает, что
При ипохондрическом
бреде больной считает, что
а) у него тяжелая болезнь
б) его обворовывают
в) он преступник
г) его хотят отравить
675. Расстройства восприятия — это
а) судороги
б) галлюцинации
в) бред
г) депрессия
676. Дромомания (влечение к бродяжничеству) — это расстройство
а) памяти
б) эмоциональной сферы
в) волевой сферы
г) депрессия
677. Основой слабоумия являются
а) грубые органические изменения клеток головного мозга
б) функциональные расстройства высшей нервной деятельности под влиянием внешних факторов (стресс)
в) стойкая дисгармония эмоционально-волевых сторон психики
г) хроническая соматическая патология
678. Деменция — это
Деменция — это
а) острый психоз
б) тоскливое, «угнетенное» настроение
в) слабоумие, приобретенное в процессе болезни
г) врожденное слабоумие
679. Галлюцинации — это
а) чувственное восприятие при отсутствии соответствующего внешнего объекта
б) искаженное восприятие реально существующего раздражителя
в) окружающие предметы видятся удвоенными
г) выпадение половины поля зрения
680. Изменение дыхания в первой фазе большого судорожного припадка
а) Чейна-Стокса
б) отсутствует
в) учащено
г) Биотта
681. Показанием для госпитализации в психиатрическую больницу является
а) маниакальное состояние без склонности к агрессии
б) антиобщественное поведение психически больного
в) неврозы
г) врожденное слабоумие
682. Состояние выключения сознания
Состояние выключения сознания
а) кома
б) делирий
в) сумеречное помрачение сознания
г) онейроид
683. Признак сумеречного помрачения сознания
а) кататоническая заторможенность
б) недоступность контакту и социально опасные действия
в) чрезмерная сонливость
г) слабая реакция на окружающие раздражители
684. Препарат для купирования некоторых видов возбуждения – раствор
а) 0,5% седуксена
б) 1% димедрола
в) 20% натрия оксибутирата
г) 50% анальгина
685. Кожа не выполняет функцию
а) защитную
б) дыхательную
в) гормональную
г) секреторную
686. К
воспалительным пятнам относятся
К
воспалительным пятнам относятся
а) эритемы
б) пурпуры
в) петехии
г) экхимозы
687. Небольшое скопление жидкости в эпидермисе или между эпидермисом и дермой
а) пятно
б) узелок
в) везикула
г) волдырь
688. Первичный морфологический элемент
а) рубец
б) лихенизация
в) трещина
г) пустула
689. Апокриновые потовые железы отсутствуют
а) в подмышечных впадинах
б) на ладонях
в) вокруг сосков молочной железы
г) в области гениталий
690. Вторичный морфологический элемент
высыпаний
Вторичный морфологический элемент
высыпаний
а) узелок
б) бугорок
в) петехия
г) корка
691. Источник заражения микроспорией (возбудитель микроспорум ржавый)
а) больной человек
б) кошки
в) крупный рогатый скот
г) собаки
692. К заразным заболеваниям относятся
а) розовый лишай
б) эритразма
в) чесотка
г) экзема
693. Наиболее частые места локализации при чесотке у взрослых
а) ладони и подошвы
б) слизистые оболочки
в) межпальцевые складки кистей
г) лицо
694. Первичный элемент при пиодермитах
Первичный элемент при пиодермитах
а) пустула
б) везикула
в) бугорок
695. Преимущественная локализация элементов при сикозе
а) лоб
б) живот
в) борода, усы
г) спина
696. Кожное заболевание, передаваемое через обувь
а) токсидермия
б) эпидермофития
в) микроспория
г) нейродермит
697. Из детских учреждений следует обязательно изолировать детей с кожным заболеванием
а) бородавками
б) микроспорией
в) истинной экземой
г) крапивницей
698. Морфологический
элемент, характерный для крапивницы
Морфологический
элемент, характерный для крапивницы
а) волдырь
б) пузырь
в) бугорок
г) пустула
699. Источник заражения при пушистой микроспории
а) крупный рогатый скот
б) кошки
в) больной человек
г) вши
700. Приоритетная проблема при чесотке
а) зуд
б) боли в мышцах
в) лихорадка
г) облысение
701. Возбудитель чесотки
а) вирусы
б) клещи
в) грибы
г) простейшие
702. Воспаление потовых желез
а) фурункул
б) лимфаденит
в) гидраденит
г) сикоз
703. Для простого
герпеса наиболее характерны
Для простого
герпеса наиболее характерны
а) лихорадка, лимфаденит
б) узелки, чешуйки, гиперпигментация
в) пузырьки, эрозии, корки
г) опоясывающие боли, зуд
704. Для стафилодермий не характерна
а) локализация пустул в волосяных фолликулах и железах
б) коническая или шаровидная форма пустул
в) напряженная покрышка пустул, развитие фолликулитов
г) локализация пустул в складках кожи
705. Инкубационный период при чесотке
а) 6 недель
б) 6 часов
в) 3 месяца
г) 6-10 дней
706. Повторный опоясывающий лишай у лиц молодого возраста подозрителен на
а) вирусные гепатиты
б) ВИЧ-инфекцию
в) сахарный диабет
г) лейкоз
707. При опоясывающем
лишае характерный симптом
При опоясывающем
лишае характерный симптом
а) разлитая гиперемия кожных покровов
б) невралгические боли
в) зуд в ночное время
г) отрубевидное шелушение
708. Ограниченную токсикодермию часто вызывает применение
а) антибиотиков
б) кортикостероидных гормонов
в) аскорбиновой кислоты
г) сульфаниламидов
709. Для диагностики микроспории волосистой части головы не применяют
а) микроскопические исследования волос из очагов поражения
б) культуральное исследование чешуек волос
в) люминесцентное исследование с помощью лампы Вуда
г) исследование с помощью йода (йодная проба)
710. Грозное осложнение при крапивнице
Грозное осложнение при крапивнице
а) зуд
б) жжение
в) головная боль
г) асфиксия
711. В целях ранней диагностики микроспории у взрослых применяют
а) УФО
б) лампу Вуда
в) лампу Соллюкс
г) УЗИ
712. Для этиотропного лечения чесотки применяют
а) димедроловую мазь
б) левомиколь
в) фреднизолон
г) эмульсию бензил-бензоата
713. При этиотропном лечении чесотки применяется
а) дерматоловая мазь
б) эмульсия синтомицина, лосьон «Ниттифор»
в) ихтиоловая мазь
г) медифокс
714. Для лечения
лобкового педикулеза применяется
Для лечения
лобкового педикулеза применяется
а) дерматоловая мазь
б) гелиомициновая мазь
в) преднизолоновая мазь
г) эмульсия бензил-бензоата
715. Потенциальная проблема при чесотке
а) пиодермия
б) экзема
в) отек Квинке
г) токсикодермия
716. Камерную дезинфекцию используют при
а) псориазе
б) головном педикулезе
в) розовом лишае
г) чесотке
717. Психологическая проблема у больных с вульгарным псориазом
а) депрессия
б) бессоница
в) угроза потери работы
г) зуд
718. Для диагностики
сифилиса используют реакцию
Для диагностики
сифилиса используют реакцию
а) Райта
б) РНГА риккетсиями Провагена
в) агглютинации
г) Вассермана
719. Основный путь заражения сифилисом
а) воздушно-капельный
б) бытовой
в) контактный
г) половой
720. Атипичные формы твердого шанкра могут быть в виде
а) язвы
б) эрозии
в) индуративного отека
г) вегетации
721. Сифилис на латинском языке
а) Pestis
б) Scabies
в) Lues
г) Lupus
722. Для вторичного
свежего сифилиса характерно появление
Для вторичного
свежего сифилиса характерно появление
а) язвы
б) эрозии
в) шанкра-амигдалита
г) папул
723. Для вторичного рецидивного сифилиса характерно появление
а) лихенификации
б) сифилитической лейкодермы
в) язвы
г) эрозии
724. Вторичный период сифилиса характеризуется появлением на коже
а) эрозии или язвы
б) пятнистых и папулезных высыпаний
в) бугорковых высыпаний
г) узлов
725. Продолжительность вторичного периода сифилиса
а) 2-4 года
б) 6 месяцев
в) 6 недель
г) 7 лет
726. Психологические
проблемы у больных венерическими
заболеваниями
Психологические
проблемы у больных венерическими
заболеваниями
а) жжение
б) отчаяние
в) угроза потери работы
г) зуд
727. Потенциальная физиологическая проблема у больного гонореей
а) бесплодие
б) отчаяние
в) депрессия
г) страх
728. Возбудитель гонореи
а) нейсерия
б) риккетсия
в) боррелия
г) спирохета
729. Для диагностики гонореи прежде всего используют
а) микроскопию
б) бактериологический посев
в) биохимические исследования
г) реакцию Кана
730. У больных
гонореей нарушается физиологическая
потребность
У больных
гонореей нарушается физиологическая
потребность
а) выделять
б) дышать
в) общаться
г) есть, пить
731. В первичном периоде сифилиса у больных не встречаются признаки
а) твердый шанкр
б) отрицательная реакция Вассермана
в) регионарный лимфаденит
г) розеолы на коже туловища
732. Инкубационный период при гонорее
а) 5–7 дней
б) 5–7 недель
в) 2 месяца
г) 6–10 месяцев
733. Острота зрения определяется при помощи
а) периметра
б) таблицы Рабкина Е.Б.
в) таблицы Сивцева Д.А.
г) рефрактометра
734. За норму принята острота зрения, равная
а) 0,5
б) 0,8
в) 0,9
г) 1,0
735. С помощью полихроматических таблиц проверяют
а) поле зрения
б) цветоощущение
в) светоощущение
г) остроту зрения
736. Помутнение хрусталика называется
а) микрофакией
б) катарактой
в) макрофакией
г) миопией
737. Характерная жалоба при зрелой катаракте
а) отсутствие предметного зрения
б) выделения из глаза
в) улучшение ранее сниженного зрения
г) боль в глазу
738. Воспаление слизистой оболочки глаза называется
а) дакриоциститом
б) конъюнктивитом
в) дакриоаденитом
г) блефаритом
739. Характер отделяемого из глаз при дифтерийном конъюнктивите
а) мутное с хлопьями
б) слизисто-гнойное, гнойное
в) цвета мясных помоев
г) отделяемое отсутствует
740. Инфильтрат роговицы – симптом
а) конъюнктивита
б) глаукомы
в) кератита
г) катаракты
741. От воздействия на глаз рентгеновских лучей может возникнуть
а) электроофтальмия
б) катаракта
в) ячмень
г) глаукома
742. Гонобленнорея новорожденного, если заражение произошло при прохождении ребенка через родовые пути, начинается после рождения
а) на 5-й день
б) через 2-3 дня
в) сразу
г) через 2 недели
743. Для профилактики гонобленнореи новорожденным закапывают в глаза раствор
а) 0,25% левомицетина
б) 30% сульфацил-натрия
в) 3% колларгола
г) фурацилина 1:5000
744. Повязку на глаз накладывают при
а) конъюнктивите
б) кератите
в) ранении глаза
г) блефарите
745. К заболеваниям век относятся
Тесты по неврологии в медицинском колледже
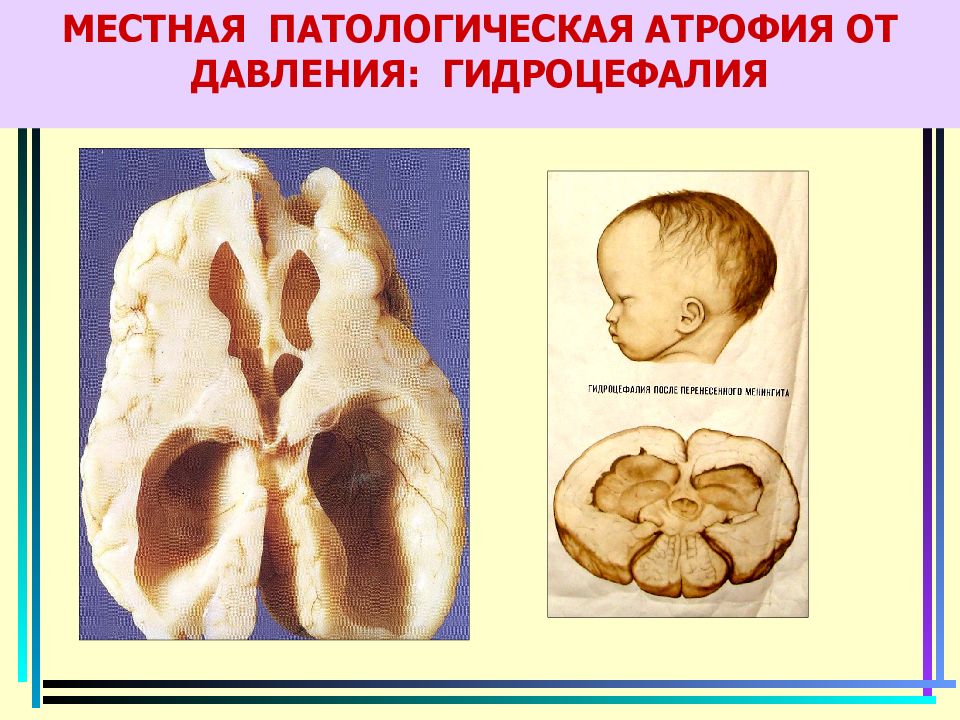
Образец ткани опухоли головного мозга
№ 2
* 1 -один правильный ответ
Менингиальный симптом — это
1) симптом Бабинского
2) ригидность мышц затылка
3) симптом «свисающей головы»
4) симптом Чураева
! 2
Смотрите также:
— Шпаргалки по неврологии
— Тесты по невропатологии для фельдшеров
— тесты по неврологии I и II вариант
№ 3
* 1 -один правильный ответ
Сознание больного при геморрагическом инсульте
1) сохранено
2) утрачено на короткое время
3) утрачено на длительное время (ком1)
4) изменено по типу сумеречного
! 3
№ 4
* 1 -один правильный ответ
При транспортировке больного с геморрагией в мозг необходимо
1) придать голове возвышенное положение
2) приподнять ноги
3) часто менять положение головы и туловища
4) избегать изменений положения головы
! 4
№ 5
* 1 -один правильный ответ
Приступообразные боли в одной половине лица, иногда со слезотечением, выделением слизи из носа, слюнотечением, возникают при
1) неврите лицевого нерва
2) невралгии тройничного нерва
3) шейном остеохондрозе
4) опухоли головного мозга
! 2
№ 6
* 1 -один правильный ответ
Симптом «заячий глаз», сглаженность лобных и носогубной складок на пораженной стороне, перекос рта в здоровую сторону характерны для
1) опухоли головного мозга
2) энцефалита
3) неврита лицевого нерва
4) острого нарушения мозгового кровообращения
! 3
№ 7
* 1 -один правильный ответ
Для спастического паралича характерно
1) снижение сухожильных рефлексов
2) атрофия мышц
3) наличие патологических рефлексов
4) снижение мышечного тонуса
! 3
№ 8
* 1 -один правильный ответ
Для периферического (вялого) паралича характерно
1) повышение сухожильных рефлексов
2) наличие патологических рефлексов
3) атрофия мышц
4) повышение мышечного тонуса
! 3
№ 9
* 1 -один правильный ответ
Спастический гемипарез — это нарушение двигательной функции в
1) обеих ногах
2) одной руке
3) одной ноге
4) руке и ноге с одной стороны
! 4
№ 10
* 1 -один правильный ответ
Невралгия — это
1) воспаление нерва
2) повреждение нерва
3) боль по ходу нерва
4) атрофия нерва
! 3
№ 12
* 1 -один правильный ответ
Клинический симптом пояснично-крестцового радикулита
1) ригидность затылочных мышц
2) симптом Ласега
3) симптом Горнера
4) симптом Брудзинского
! 2
№ 13
* 1 -один правильный ответ
Доказательное исследование, позволяющее поставить диагноз менингита
1) увеличение СОЭ крови
2) лейкоцитоз крови
3) изменение ликвора
4) лимфопения крови
! 3
№ 14
* 1 -один правильный ответ
Основной симптом миастении
1) судороги
2) мышечная утомляемость
3) головная боль
4) тремор кистей
! 2
№ 15
* 1 -один правильный ответ
Для болезни Паркинсона характерно
1) умственная деградация
2) нарушение координации
3) тремор кистей
4) параличи конечностей
! 3
№ 16
* 1 -один правильный ответ
При геморрагическом инсульте необходимо
1) придать больному положение с опущенным головным концом
2) приложить на голову пузырь со льдом
3) повернуть больного на бок
4) придать больному полусидячее положение
! 2
№ 17
* 1 -один правильный ответ
Нарастающие, упорные головные боли распирающего характера и явления застоя на глазном дне характерны для
1) энцефалита
2) менингита
3) опухоли головного мозга
4) рассеянного склероза
! 3
№ 18
* 1 -один правильный ответ
Характерный признак невралгии тройничного нерва
1) рвота
2) отсутствие складок на лбу при поднимании бровей
3) приступообразные боли в одной половине лица
4) ригидность затылочных мышц
! 3
№ 19
* 1 -один правильный ответ
Симптом «заячий глаз» бывает при
1) опухоли головного мозга
2) энцефалите
3) неврите лицевого нерва
4) остром нарушении мозгового кровообращения
! 3
№ 20
* 1 -один правильный ответ
Аура характерна для
1) геморрагического инсульта
2) менингита
3) энцефалита
4) эпилепсии
! 4
№ 21
* 1 -один правильный ответ
Основной симптом тяжелого сотрясения головного мозга
1) головная боль
2) шум в ушах
3) ретроградная амнезия
4) головокружение
! 3
№ 22
* 1 -один правильный ответ
Атрофия мышц — это основной симптом
1) спастического паралича
2) вялого паралича
3) миастении
4) болезни Паркинсона
! 2
№ 23
* 1 -один правильный ответ
Ощущение «треска» в шее при поворотах головы характерно для
1) менингита
2) остеохондроза
3) энцефалита
4) опухоли головного мозга
! 2
№ 24
* 1 -один правильный ответ
Патологические рефлексы характерны для
1) вялого паралича
2) спастического паралича
3) менингита
4) энцефалита
! 2
№ 25
* 1 -один правильный ответ
Выпячивание родничка у грудных детей наблюдается при
1) менингите
2) энцефалите
3) эпилепсии
4) полиомиелите
! 1
Эффективное лечение атрофии мышц в Москве на DocDoc.ru
Неврологи Москвы — последние отзывы
Доктора вызывали на дом. Меня удовлетворил приём, видно что она грамотный, квалифицированный специалист, я верю что она нам поможет разобраться с диагнозом моей мамы. Она провела осмотр, назначала анализы. Общалась хорошо, умело подбирает анамнез, профессионал, умеет общаться с больными и родственниками. Рекомендую ее, так как сейчас сложно найти грамотного специалиста. .
Наталья, 11 августа 2021
Прием мне очень понравился, прям вышла оттуда с другим настроением. Врач много мне чего рассказала, объяснила, уточняла, внимательно меня выслушала, назначила препарат более слабый чем до этого, что мне нравится. Так что я осталась очень довольна. Порекомендую доктора знакомым.
Ярослава, 11 августа 2021
На приеме доктор опросил меня по поводу моей проблемы, объяснил какие причины, назначил анализы и предварительно выписал лечение и препараты. Пойду повторно после получения анализов. Доктор все доходчиво объясняет, впечатления приятные остались после приема.
На модерации, 11 августа 2021
Доктор на приеме внимательно меня осмотрел, задал все вопросы про возможные прошедшие заболевания, дал мне ряд рекомендаций и назначил лечения, выбрал мягкий вариант лечения, чтобы не перезагружать организм. Наталья Николаевна подробно все объясняет. Единственный минус это дорогое лечение со стороны клиники.
Алексей, 10 августа 2021
Прежде всего хочу поблагодарить за двойную запись к специалистам. Прием у Вячеслава Юрьевича прошел достаточно быстро, профессионально и даже раньше меня принял, посмотрел все мои анализы, сделал все, что было в его силах и он меня устроил. Очень профессиональный, внимательный, хороший врач, добродушный, отзывчивый, вникающий полностью в проблему.
Филипп, 10 августа 2021
Опытный невролог и прекрасная женщина. Прием ведет размерено, внимательно выслушивает стенания пациента, ни разу ни прерывает. Мою проблему взяла под контроль, лечение назначенное помогает. Потраченных денег не жаль.
Ксения, 09 августа 2021
Профессионал своего дела. Доктор выявила причину моих болей, назначила лечение. Я еще там сделала УЗИ очень качественно, сразу определили проблему. Я очень довольна. Порекомендовала бы врача знакомым.
Эльвира, 05 августа 2021
Хороший доктор. Планирую её посетить повторно. Врач мне понравилась как с профессиональной, так и с человеческой точки зрения. Она внимательная, не навязчивая, всё понятно и доступно объясняет. Могу рекомендовать данного специалиста своим знакомым, если потребуется. Качеством приёма я остался доволен.
Вячеслав, 27 июля 2021
Специалист некомпетентен. Во время приема отвечала на звонки личного телефона. Задавала Очень поверхностные вопросы касаемо моей проблемы. Пожалуйста, просьба к таким «специалистам» , если вы не занимаетесь каким либо вопросом или плохо осведомлены в курсах лечения и консультации, пожалуйста, внесите поправки в своё резюме, уберите ненужное.
Елена, 27 июля 2021
Доктор хорошо ответила на мои вопросы и назначила необходимые обследования. Врач профессиональный. Она помогла! Такого специалиста редко можно найти.
Гурун, 18 июня 2021
Показать 10 отзывов из 15027Миопатия: причины, симптомы, лечение. в Солнцево
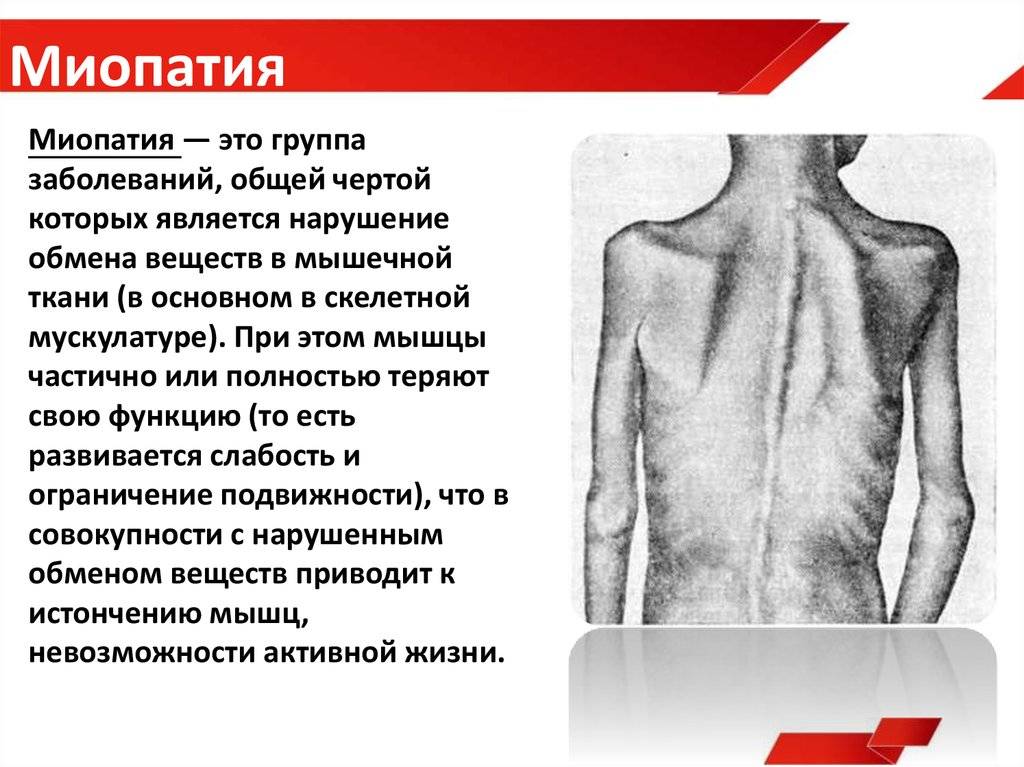
Миопатией называют совокупность симптомов, возникающих в результате повреждения мышц. В отличие от заболеваний периферической нервной системы, (при которых мышцы также повреждены, но это связано с предшествующим повреждением снабжающих их нервов), при миопатиях болезненный процесс локализуется в самой мышце.
Беспокоит слабость в конечностях? Записывайтесь к неврологу!
Миопатии представляют собой очень большую группу заболеваний с различными причинами, разным течением и прогнозом. Общей чертой является мышечная слабость — поражение мышц бедер и рук, хотя мышцы лица также могут быть ослаблены. Сенсорные расстройства при миопатиях не встречаются. Мышечная слабость обычно двусторонняя и симметричная с самого начала, то есть сходна с обеих сторон тела — левой и правой.Миопатии могут быть генетическими или приобретенными. Генетически обусловленные миопатии включают, например, мышечные дистрофии, которые характеризуются аномальной структурой мышечных клеток (например, из-за врожденного недостатка любого из его компонентов). С другой стороны, приобретенные миопатии имеют воспаление (воспалительные миопатии), могут сопровождать эндокринные заболевания (например, гипотиреоз), а также могут возникать в результате повреждения мышц некоторыми лекарственными средствами или токсичными соединениями (например, алкоголем).
Насколько распространена миопатия?
Термин «миопатия» очень широк и охватывает множество различных заболеваний. Поэтому общую заболеваемость миопатией трудно определить. Как правило, миопатии являются редкими заболеваниями.
Как проявляется миопатия?
Основным симптомом миопатии является мышечная слабость. Чаще всего это касается мышц бедер и рук — пациентам, например, трудно подниматься по лестнице, вставать из положения сидя, сидеть или выполнять действия с поднятыми руками (например, повесить шторы или делать прическу). Мышечная слабость может сопровождаться болью. При некоторых миопатиях участвуют мышцы лица или горла. Может быть опущение век, снижение подвижности глазных яблок, невнятная речь, затрудненное глотание. Иногда мышцы шеи слабые — тогда не держится голова. При некоторых миопатиях может возникнуть слабость дыхательных мышц, приводящая к одышке. Иногда при миопатиях наблюдаются проблемы с расслаблением мышц, например, пациент не может быстро разжать пальцы после сжимания кулака. При прогрессирующих и длительных миопатиях возникает атрофия мышц. Эти симптомы могут сопровождаться признаками повреждения других тканей и органов вне мышечной системы.
Что делать, если появились симптомы миопатии (слабости в конечностях)?
Если замечены какие-либо симптомы, которые могут указывать на миопатию, нужно обратиться к врачу общей практики или терапевту, который затем направит пациента к специалисту, обычно неврологу. Если симптомы длятся недолго (к примеру, несколько недель) и быстро нарастают, следует как можно скорее прийти к врачу.Как врач диагностирует миопатию?
Врач собирает историю болезни пациента и проводит тест, который обычно показывает парез мышц (бедра, руки, иногда лица), иногда также ослабление рефлексов, вызванных при постукивании с помощью неврологического молотка. Затем он заказывает дополнительные тесты. Обычно это лабораторные анализы крови, в том числе уровни электролита, ТТГ, воспаления и креатинкиназы. Это чувствительный показатель разрушения мышц, и его уровень повышается у большинства пациентов с миопатией. Он также может быть увеличен при заболеваниях, когда повреждение мышц вызвано повреждением нервов, таких как боковой амиотрофический склероз или полинейропатия, а иногда и у здоровых людей, например, после физической нагрузки или травмы мышц. Электромиография (ЭМГ) является еще одним дополнительным тестом, важным для диагностики. ЭМГ позволяет подтвердить миопатию или указывает на другое заболевание, которое вызывает аналогичные симптомы. Окончательный диагноз миопатии, то есть определение ее типа и причины, чаще всего возможен после теста среза мышечной ткани (для этой цели берут маленький фрагмент ткани под местной анестезией для микроскопического исследования) или генетического анализа из образца крови, взятого у пациента. Генетическое тестирование проводится только для некоторых генетических миопатий.
Как лечить миопатию?
Как и в случае с полиневропатией, можно выделить причинно-следственное и симптоматическое лечение. Возможно причинное лечение, например, при воспалительных миопатиях. В этих случаях используются глюкокортикоиды (стероидные гормоны), иммунодепрессанты или внутривенные иммуноглобулины. Причинное лечение миопатии, возникающей при эндокринных заболеваниях (например, заболевание щитовидной железы, гиперпаратиреоз или гиперадренокортицизм), включает лечение гормональных нарушений. При лекарственной или токсической миопатии нужно попытаться перестать принимать лекарство или прекратить воздействие токсического соединения, которое вызвало повреждение мышц. В редких случаях генетической миопатии, суть которой заключается в отсутствии одного из ферментов, необходимых для правильного функционирования мышц, возможно причинное лечение в виде введения недостающего фермента.
Симптоматическое лечение — это прежде всего реабилитация. В случае, если для пациента с быстрым мышечным расслаблением возникают обременительные трудности, фармакологическое лечение используется для смягчения этого симптома. При некоторых заболеваниях мышц поражаются другие органы, такие как сердечная мышца. Тогда для пациентов важно регулярно проходить осмотры у кардиолога. Кроме того, генетическое консультирование следует давать пациентам с генетической миопатией.
Можно ли полностью вылечить миопатию?
При генетических миопатиях полное выздоровление невозможно. Прогноз для приобретенных миопатий зависит от их причины: например, при воспалительных миопатиях мышечная сила обычно улучшается после начала лечения, а в миопатии, вызванной гормональными нарушениями, мышечные симптомы обычно исчезают после излечения основного заболевания.
Обращайтесь в поликлинику Медсемья в Солнцево — наш врач-невролог проведет диагностику и назначит правильное лечение!
Лечение мышечной атрофии | медцентр Аватаж
Суть процесса мышечной атрофии состоит в уменьшении объема мышц и перерождении мышечных волокон. Они истончаются, деформируются, впоследствии их число может существенно сократиться вплоть до полного исчезновения. Вследствие атрофии мышц мышечная ткань постепенно заменяется на соединительную ткань, которая неспособна выполнять функцию движения. Происходит утрата мышечной силы, снижается мышечный тонус, что вспоследствии приводит к частичной или полной обездвиженности пораженных групп мышц.
Существует две основные формы заболевания: первичная и вторичная.
Первичная мышечная атрофия диагностируется при выявлении признаков поражения самой мышечной ткани. Как правило, в данном случае речь идет о наследственных болезнях (так называемых миопатиях). При этих заболеваниях обнаруживают врожденный дефект ферментов мышц либо высокую проницаемость мембран клеток-миоцитов. Очень часто болезнь дебютирует в детском и юношеском возрасте, однако встречаются и иные формы, которые проявляются в 30-40 лет и даже позднее. Спровоцировать реализацию генетического дефекта мышц или самостоятельно стать причиной атрофии могут внешние факторы: травмы, нарушения кровообращения, физические перегрузки, инфекционные болезни.
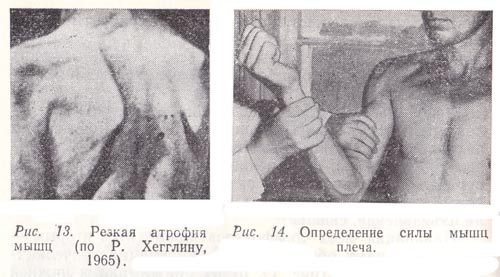
Атрофия мышц
Вторичная (или невральная) атрофия является следствием поражения не мышечной, а нервной ткани. Вследствие разных причин могут страдать периферические нервы, корешки, передние рога спинного мозга. Часто процесс развивается на фоне злокачественных новообразований, травм нервных стволов или спинного мозга, воздействия токсических веществ и инфекционных агентов (например, вируса полиомиелита), нарушений обмена веществ (в частности, при голодании, старении организма), длительной двигательной бездеятельности. Существуют также и наследственные заболевания, основным проявлением которых является вторичная мышечная атрофия.
Клиника мышечной атрофииКлиника мышечных атрофий весьма разнообразна. Как первичная, так и вторичная атрофии имеют множество форм, каждая из которых по сути представляет собой самостоятельное заболевание со своей симптоматикой, течением и прогнозом. В одних случаях инвалидизация пациента может наступить уже на протяжении нескольких месяцев, в других – больные могут сохранять двигательные функции десятилетиями. Общими признаками всех атрофий является заметное уменьшение объема пораженных мышц, снижение ее тонуса, та или иная степень двигательного дефекта, боли при пальпации. Могут наблюдаться подергивания в мышце, нарушения чувствительности, снижение или исчезновение сухожильных рефлексов. В некоторых случаях болезнь начинается с дистальных групп мышц (более отдаленных от туловища) – голень, стопа, кисти рук, в других же – с проксимальных (более приближенных туловищу) – мышцы бедра и плечевого пояса. Врачам-клиницистам хорошо знакомы внешние признаки мышечных атрофий, позволяющие поставить пациенту предварительный диагноз уже во время осмотра: «утиная походка» (больной ходит как утка, переваливаясь с ноги на ногу), «обезьянья лапа» (специфическая форма кисти при атрофии межпальцевых мышц), «симптом лестницы» (при вставании из сидячего положения больной помогает себе руками) и другие.
Диагностика мышечной атрофии
Диагностика мышечных атрофий включает в себя обязательный осмотр врача-невролога с тщательной оценкой неврологического статуса. Из дополнительных методов обследования используют электромиографию (ЭМГ), компьютерную и магнитно-резонансную томографию. Иногда приходится прибегать к специфическим анализам крови (например, оценке иммунологических показателей для исключения инфекционного поражения нервной системы) и спинномозговой пункции.
Мышцы человека
Лечение мышечной атрофии
Лечение мышечных атрофий – крайне сложный комплексный процесс. Он включает в себя консервативную медикаментозную терапию, а также ряд нефармакологических методик. При возникновении контрактур их устраняют редрессациями, с дальнейшей фиксацией гипсовыми повязками. Особо стоит отметить большое значение правильного питания, лечебной физкультуры, массажа, психотерапии, а также физиотерапевтических процедур.
Правильно подобранное лечение позволяет замедлить темпы прогрессирования атрофий и сохранить работоспособность больных на долгие годы.
Ударно-волновая терапия (УВТ) применяется в лечении мышечных атрофий относительно недолго, однако метод уже успел завоевать доверие европейских врачей и пациентов благодаря своей высокой эффективности, безопасности и широкими возможностями комплексного применения.
Ударные волны оказывают выраженное положительное воздействие на мышечную ткань благодаря улучшению ее кровоснабжения и трофики, что в свою очередь способствует регенерации и росту мышечных волокон. Улучшение метаболизма и усвоения кислорода мышцей приводит к постепенным структурным изменениям. Разрыхляется и рассасывается соединительная фиброзная ткань, которая не выполняет двигательную функцию. Происходит рост кровеносных сосудов в мышце (неоангиогенез), что также благоприятно сказывается на ее питании. Все вышеперечисленное позволяет добиться хорошего двигательного эффекта и улучшения качества жизни пациента.
Лечение мышечной атрофии ударно-волновой терапией Курс лечения УВТ – до 10 процедур. В лечении мышечных атрофий в клинике Аватаж кроме ударных волн активно используется технология V-Aactor, что дополнительно улучшает микроциркуляцию и лимфоотток в пораженных мышцах. Сеанс ударно-волновой терапии занимает до 30 минут, не требует никакой предварительной подготовки. Интервал между процедурами – не менее 1 недели. УВТ применяется в комплексе со стандартным медикаментозным лечением и лечебной гимнастикой. Эффект от применения метода нарастает и после окончания курса лечения.
Автор: Куц К.В.
Врач невролог клиники Аватаж
Разрыв ахиллова сухожилия — цены на лечение, симптомы и диагностика заболевания в клинике «Мать и дитя» в Перми
Симптомы разрыва ахиллова сухожилия
К основным признакам повреждения сухожилия относят:
- Острую боль. Она возникает непосредственно в области повреждения и сравнивается пациентами с ощущениями от пореза или резкого удара. В некоторых случаях боль является плохо переносимой и требует немедленной госпитализации больного.
- Отечность. Она возникает непосредственно в области сухожилия, но затем может распространиться по всей голени.
- Ограничения в подвижности. Пациент не может встать на носочки или согнуть стопу в сторону подошвы.
При прощупывании задней поверхности голени специалист может выявить «провал». Обычно он располагается чуть выше места прикрепления к пяточной кости ахилла.
Если повреждение является застарелым, то к основным симптомам присоединяется уменьшение объема больной голени, что связано с атрофией мышц.
Причины разрыва ахиллова сухожилия
Самой распространенной причиной разрыва сухожилия является прямая травма. Повреждение является следствием удара палкой, ногой и др. Также сухожилие может разорваться из-за резкого сокращения мышцы. Обычно травмы возникают во время начала бега или вследствие чрезмерного сгибания стопы при падении. Вероятность повреждения существенно повышается в случае, если физическая нагрузка оказывается без предварительного разогрева мышцы. Именно поэтому перед любыми спортивными занятиями или существенными физическими нагрузками рекомендуют выполнять хотя бы самые простые упражнения, позволяющие подготовить опорно-двигательный аппарат.
Диагностика разрыва ахиллова сухожилия в клинике
Диагноз может быть поставлен уже на основании клинических симптомов врачом-травматологом. Опытный специалист сразу же видит клиническую картину травмы и может определить место повреждения опорно-двигательного аппарата. Пациенту следует быть готовым ответить на поставленные врачом вопросы о перенесенной травме, симптомах, времени, которое прошло с момента повреждения и др.
Тем не менее, в ряде ситуаций может потребоваться и комплексное обследование пациента. В нашей клинике оно проводится с использованием стандартных и современных методик. Обследования не занимают много времени и позволяют поставить точный диагноз. Для точной оценки применяется современное оборудование экспертного класса, которое отличается высокой степенью визуализации.
Способы обследования
- Рентгенография.
- УЗИ.
- МРТ.
Все эти обследования назначаются для уточнения распространенности повреждения, а также выявления выраженности патологических изменений сухожилия. Диагностика не занимает много времени.
Лечение разрыва ахиллова сухожилия в клинике
При разрыве появляется диастаз (определенное расстояние) между поврежденными концами сухожилия. Это приводит к тому, что естественное восстановление становится невозможным, как и консервативная терапия. Проводится исключительно оперативное лечение.
При свежих разрывах возможно использование методик, не подразумевающих разреза. Сухожилие сшивается через кожу. Вмешательство занимает 30-60 минут и легко переносится пациентом. Использование общего наркоза не является обязательным. После выполнения операции пациенту на 1 месяц накладывают гипсовую повязку. Затем швы снимаются, а гипс накладывается еще раз. Если разрыв является застарелым, повреждение ушивается открытым способом. Дополнительно проводится пластика сухожилия.
В некоторых случаях дополнительно производится укрепление ахилла. Оно актуально для спортсменов и всех остальных людей, которые часто подвергаются повышенным физическим нагрузкам. Для такого вмешательства используют фасцию бедра (соединительнотканную оболочку) пациента. После ушивания гипс накладывают на 1,5 месяца. Пациенту при этом разрешают передвигаться сразу же (обязательно с дополнительной опорой). Также назначается лечебная физкультура. Полноценная нагрузка на ногу обеспечивается спустя 2 месяца.
Важно! Выбор в пользу методики выполнения операции делает исключительно врач. При этом он ориентируется на имеющиеся показания и противопоказания. Учитываются срок получения травмы и все ее особенности, а также индивидуальные параметры пациента и другие факторы.
Профилактика разрыва ахиллова сухожилия и врачебные рекомендации
Чтобы предотвратить вероятность получения травмы, рекомендуют:
- Отказываться от любых физических занятий при появлении даже несущественной боли в области ахиллова сухожилия.
- Носить только удобную и правильную обувь. При использовании моделей с гибкой подошвой можно сократить вероятность травмирования.
- Обращаться к врачу при любом дискомфорте в области пятки и в зоне немного выше нее.
- Выполнять простые упражнения на растяжку мышц и в целом области ахиллова сухожилия.
- Проводить интенсивные тренировки с использованием эластичных бинтов или специальных ортезов. Это позволяет снизить нагрузку на сухожилие и предотвратить риски его травмирования.
Важно! Любые упражнения рекомендуется выполнять под контролем опытного специалиста. Он снизит вероятность травмы и позволит обеспечить дозированные нагрузки.
Если вы планируете обратиться к врачу для профилактики разрывов и иных повреждений ахиллова сухожилия, планируете проведение оперативного вмешательства в нашей клинике после полученной травмы, позвоните по указанному на сайте номеру или отправьте заявку через форму обратной связи. Наш специалист ответит на все ваши вопросы и запишет на консультацию в удобное время.
Каковы признаки и симптомы очаговой мышечной атрофии (ФМА)?
Юбельт Б., Друкер Дж. Постполиомиелитный синдром: обновленная информация. Семин Нейрол . 1993 сентября, 13 (3): 283-90. [Медлайн].
Ramlow J, Alexander M, LaPorte R, Kaufmann C, Kuller L. Эпидемиология постполиомиелитного синдрома. Am J Epidemiol . 1992 г., 1. 136 (7): 769-86. [Медлайн].
Альстром Г., Гуннарссон Л.Г., Лейсснер П., Сьоден П.О. Эпидемиология нервно-мышечных заболеваний, включая последствия после полиомиелита, в шведском округе. Нейроэпидемиология . 1993. 12 (5): 262-9. [Медлайн].
Такемура Дж., Саэки С., Хатисука К., Аритоме К. Распространенность постполиомиелитного синдрома на основе поперечного исследования в Китакюсю, Япония. J Rehabil Med . 2004 г., 36 (1): 1-3. [Медлайн].
Ивани Б., Ноллет Ф., Редекоп В.К. и др. Последствия полиомиелита с поздним началом: инвалидность и увечья в популяционной когорте вспышки полиомиелита 1956 г. в Нидерландах. Arch Phys Med Rehabil . 1999 июн. 80 (6): 687-90. [Медлайн].
Wekre LL, Stanghelle JK, Lobben B, Oyhaugen S. Норвежское исследование полиомиелита 1994: общенациональное исследование проблем давнего полиомиелита. Спинной мозг . 1998 апр. 36 (4): 280-4. [Медлайн].
Сегал Х. Новые измерения полиомиелита. Индийский педиатр . 1990 Май. 27 (5): 433-6. [Медлайн].
Четвинд Дж., Боттинг К., Хоган Д.Постполиомиелитный синдром в Новой Зеландии: опрос 700 выживших после полиомиелита. N Z Med J . 1993 22 сентября. 106 (964): 406-8. [Медлайн].
Нагашима Т. [Поздняя прогрессирующая мышечная атрофия после полиомиелита (PPMA) — клинический анализ случаев в Японии]. Риншо Синкэйгаку . 1991 31 декабря (12): 1319-21. [Медлайн].
Гурье-Деви М, Суреш Т.Г., Шанкар СК. Мономерная амиотрофия. Arch Neurol . 1984, апрель, 41 (4): 388-94. [Медлайн].
Ким Дж. Й., Ли К. В., Ро Дж. К., Чи Дж. Г., Ли С. Б.. Клиническое исследование доброкачественной очаговой амиотрофии. J Корейская медицина . 1994, апрель, 9 (2): 145-54. [Медлайн]. [Полный текст].
Saha SP, Das SK, Gangopadhyay PK, Roy TN, Maiti B. Картина болезни двигательных нейронов в восточной Индии. Acta Neurol Scand . 1997 июл.96 (1): 14-21. [Медлайн].
Конус ЛА, Наземи Р., Конус Миссури. Обратимое БАС-подобное расстройство при ВИЧ-инфекции.БАС-подобный синдром с новой ВИЧ-инфекцией и полным ответом на антиретровирусную терапию. Неврология . 13 августа 2002 г. 59 (3): 474; ответ автора 474-5. [Медлайн].
Уэяма Х., Кумамото Т., Джоно М., Мита С., Цуда Т. Локальное истощение мышц как начальный симптом лимфомы скелетных мышц. J Neurol Sci . 1998 21 января. 154 (1): 113-5. [Медлайн].
Дубовиц В., Платтс М. Заболевание центрального ядра мышцы с очаговым истощением. J Neurol Neurosurg Psychiatry . 1965 28 октября (5): 432-7. [Медлайн]. [Полный текст].
Dalakas MC, Sever JL, Madden DL и др. Поздняя постполиомиелитная мышечная атрофия: клинические, вирусологические и иммунологические исследования. Ред. Заразить Dis . 1984 май-июнь. 6 Приложение 2: S562-7. [Медлайн].
Halstead LS, Серебряный JK. Непаралитический полиомиелит и постполиомиелитный синдром. Am J Phys Med Rehabil . 2000 янв-фев. 79 (1): 13-8. [Медлайн].
Trojan DA, Collet J, Pollak MN и др. Сывороточный инсулиноподобный фактор роста-I (IGF-I) не коррелирует положительно с изометрической силой, утомляемостью и качеством жизни при постполиомиелитном синдроме. J Neurol Sci . 2001, 1. 182 (2): 107-15. [Медлайн].
Гонсалес Х, Олссон Т, Борг К. Управление постполиомиелитным синдромом. Ланцет Нейрол . 2010 июн.9 (6): 634-42. [Медлайн].
Farbu E, Rekand T, Tysnes OB, Aarli JA, Gilhus NE, Vedeler CA.Антитела GM1 при постполиомиелитном синдроме и ранее перенесенном паралитическом полиомиелите. Дж Нейроиммунол . 2003 июн. 139 (1-2): 141-4. [Медлайн].
Gonzalez H, Ottervald J, Nilsson KC, et al. Идентификация новых белковых биомаркеров-кандидатов для постполиомиелитного синдрома — значение для диагностики, нейродегенерации и нейровоспаления. Дж. Протеомика . 2009 30 января. 71 (6): 670-81. [Медлайн].
Fordyce CB, Gagne D, Jalili F, Alatab S, Arnold DL, Da Costa D.Повышенные маркеры воспаления в сыворотке крови при постполиомиелитном синдроме. J Neurol Sci . 2008 15 августа. 271 (1-2): 80-6. [Медлайн].
Остлунд Г., Валин Å, Суннерхаген К.С., Борг К. Синдром постполиомиелита: утомленные пациенты — особая подгруппа ?. J Rehabil Med . 2011 Январь 43 (1): 39-45. [Медлайн].
Gourie-Devi M, Nalini A. Долгосрочное наблюдение за 44 пациентами с плечевой мономерной амиотрофией. Acta Neurol Scand .2003 Март 107 (3): 215-20. [Медлайн].
De Freitas MR, Nascimento OJ. Доброкачественная мономерная амиотрофия: исследование двадцати одного случая. Arq Neuropsiquiatr . 2000 Сентябрь 58 (3B): 808-13. [Медлайн].
Хираяма К., Томонага М., Китано К., Ямада Т., Кодзима С., Араи К. Очаговая шейная полиопатия, вызывающая ювенильную мышечную атрофию дистального отдела верхней конечности: патологическое исследование. J Neurol Neurosurg Psychiatry . 1987 Mar.50 (3): 285-90.[Медлайн]. [Полный текст].
Морено Мартинес Дж. М., Гарсия де ла Роча М. Л., Мартин Арагуз А. [Мономерная сегментарная амиотрофия: испанский случай с поражением ноги]. Rev Neurol (Париж) . 1990. 146 (6-7): 443-5. [Медлайн].
Ориема Дж., Эшби П., Шпигель С. Мономельная атрофия. Can J Neurol Sci . 1990 Май. 17 (2): 124-30. [Медлайн].
Серратрис Г., Пеллиссье Дж. Ф., Пуже Дж. [Нозологическое исследование 25 случаев хронической мономерной амиотрофии]. Rev Neurol (Париж) . 1987. 143 (3): 201-10. [Медлайн].
Хираяма К., Токумару Ю. Шейный дуральный мешок и спинной мозг при ювенильной мышечной атрофии дистального отдела верхней конечности. Неврология . 2000 г. 23 мая. 54 (10): 1922-6. [Медлайн].
Biondi A, Dormont D, Weitzner I Jr, Bouche P, Chaine P, Bories J. MR. Визуализация шейного отдела спинного мозга при ювенильной амиотрофии дистального отдела верхней конечности. AJNR Am J Neuroradiol .1989 март-апрель. 10 (2): 263-8. [Медлайн].
Хуан Ю.Л., Чен СиДжей. Болезнь Хираямы. Клиника нейровизуализации N Am . 2011 21 ноября (4): 939-50, ix-x. [Медлайн].
Баба Ю., Накадзима М., Уцуномия Х. и др. Магнитно-резонансная томография грудного эпидурального расширения вен при болезни Хираямы. Неврология . 2004, 27 апреля. 62 (8): 1426-8. [Медлайн].
Guglielmo GD, Brahe C, Di Muzio A. Доброкачественные мономерные амиотрофии верхних и нижних конечностей не связаны с делециями гена выживания моторного нейрона. J Neurol Sci . 1996 15 сентября. 141 (1-2): 111-3. [Медлайн].
Fetoni V, Briem E, Carrara F, Mora M, Zeviani M. Мономерная амиотрофия, связанная с мутацией 7472insC в гене tRNASer (UCN) мтДНК. Нервно-мышечное расстройство . 2004 14 ноября (11): 723-6. [Медлайн].
Ито С., Кувабара С., Фукутаке Т., Токумару Ю., Хаттори Т. Гиперигемия у пациентов с ювенильной мышечной атрофией дистального отдела верхней конечности (болезнь Хираямы). J Neurol Neurosurg Psychiatry . 2005 Январь 76 (1): 132-4. [Медлайн]. [Полный текст].
Налини А., Локеш Л., Ратнавалли Э. Семейная мономерная амиотрофия: случай из Индии. J Neurol Sci . 2004 15 мая. 220 (1-2): 95-8. [Медлайн].
Серратрис Г. Фокальные формы денервационных расстройств. Прогресс в клинической неврологии Под ред. Синха К.К., Чандра П., Neurological Soci . 1990. 6 (2): 49-54.
Тандан Р., Шарма К.Р., Брэдли В.Г., Беван Х., Якобсен П.Хроническая сегментарная спинальная мышечная атрофия верхних конечностей у однояйцевых близнецов. Неврология . 1990 Февраль 40 (2): 236-9. [Медлайн].
Серратрис Г., По-Серрадель А., Пеллиссье Дж. Ф., Ру Н., Ламарко-Чивро Дж., Пуже Дж. Хронические нейрогенные амиотрофии четырехглавой мышцы. Дж. Neurol . 1985. 232 (3): 150-3. [Медлайн].
Веселая DE. Молекулярный патогенез спинальной и бульбарной мышечной атрофии. Мозг Res Bull . 2001 окт-1 ноя.56 (3-4): 203-7. [Медлайн].
Кацуно М., Адачи Х., Танака Ф., Собуэ Г. Спинальная и бульбарная мышечная атрофия: лиганд-зависимый патогенез и терапевтические перспективы. Дж Мол Мед (Берл) . 2004 г., май. 82 (5): 298-307. [Медлайн].
Ферранте Массачусетс, Уилборн AJ. Характерные электродиагностические особенности болезни Кеннеди. Мышечный нерв . 1997 Mar.20 (3): 323-9. [Медлайн].
Lederman RJ, Salanga VD, Wilbourn AJ, Hanson MR, Dudley AW Jr.Очаговая воспалительная миопатия. Мышечный нерв . 1984 7 февраля (2): 142-6. [Медлайн].
Лорентчук Н., Фалькенберг М.П., Пирпирис М. Первичный бактериальный пиомиозит, связанный с септическим артритом, вызванным Streptococcus pyogenes: клинический случай. Ам Дж Ортоп . 2003 марта 32 (3): 148-50. [Медлайн].
Ван Дж.Й., Ли Л.Н., Сюэ П.Р. и др. Туберкулезный миозит: редкое, но существующее клиническое заболевание. Ревматология (Оксфорд) .2003 июл.42 (7): 836-40. [Медлайн].
Chen SS, Chien CH, Yu HS. Синдром фиброзной контрактуры дельтовидной и / или ягодичной мышцы: инъекционная миопатия. Acta Neurol Scand . 1988 Сентябрь 78 (3): 167-76. [Медлайн].
Серор П. Невралгическая амиотрофия. Обновление. Костный сустав . 2017 Март 84 (2): 153-158. [Медлайн].
ван Альфен Н. Клинико-патофизиологические концепции невралгической амиотрофии. Nat Rev Neurol . 2011 10 мая. 7 (6): 315-22. [Медлайн].
van Alfen N, van Engelen BG, Hughes RA. Лечение идиопатической и наследственной невралгической амиотрофии (плечевого неврита). Кокрановская база данных Syst Rev . 8 июля 2009 г. (3):
Леман В.Т., Лютмер PH, Соренсон Э.Дж., Картер Р.Э., Гупта В., Флетчер Г.П. Результаты МРТ-визуализации шейного отдела позвоночника у пациентов с болезнью Хираямы в Северной Америке: исследование с несколькими участками. AJNR Am J Neuroradiol .2013 Февраль 34 (2): 451-6. [Медлайн].
Fleckenstein JL, Peshock RM, Lewis SF, Haller RG. Магнитно-резонансная томография повреждений и атрофии мышц при гликолитических миопатиях. Мышечный нерв . 1989, 12 октября (10): 849-55. [Медлайн].
Schwennicke A, Bargfrede M, Reimers CD. Клиническая, электромиографическая и ультразвуковая оценка очаговых невропатий. Дж. Нейровизуализация . 1998 июл.8 (3): 136-43. [Медлайн].
Миллер, округ Колумбия.Постполиомиелитная патология спинного мозга. Отчет о клиническом случае с иммунопатологом. Ann N Y Acad Sci . 1995 25 мая. 753: 186-93. [Медлайн].
Бричта Л., Холкер И., Хауг К., Клокгайд Т., Вирт Б. Активация SMN in vivo у носителей спинальной мышечной атрофии и пациентов, получавших вальпроат. Энн Нейрол . 2006 июн. 59 (6): 970-5. [Медлайн].
Peel MM, Cooke M, Lewis-Peel HJ, Lea RA, Moyle W. Рандомизированное контролируемое испытание коэнзима Q10 для лечения усталости при поздних последствиях полиомиелита. Комплемент Тер Мед . 2015 23 декабря (6): 789-93. [Медлайн].
Querin G, D’Ascenzo C, Peterle E, Ermani M, Bello L, Melacini P. Пилотное испытание кленбутерола при спинальной и бульбарной мышечной атрофии. Неврология . 2013 г. 4 июня. 80 (23): 2095-8. [Медлайн].
Fernández-Rhodes LE, Kokkinis AD, White MJ, et al. Эффективность и безопасность дутастерида у пациентов со спинальной и бульбарной мышечной атрофией: рандомизированное плацебо-контролируемое исследование. Ланцет Нейрол . 2011 10 февраля (2): 140-7. [Медлайн]. [Полный текст].
Horemans HL, Nollet F, Beelen A, et al. Пиридостигмин при постполиомиелитном синдроме: отсутствие снижения утомляемости и ограниченное функциональное улучшение. J Neurol Neurosurg Psychiatry . 2003 декабрь 74 (12): 1655-61. [Медлайн]. [Полный текст].
Vasconcelos OM, Prokhorenko OA, Salajegheh MK, Kelley KF, Livornese K, Olsen CH. Модафинил для лечения усталости при постполиомиелитном синдроме: рандомизированное контролируемое исследование. Неврология . 2007 15 мая. 68 (20): 1680-6. [Медлайн].
Купман Ф.С., Уэгаки К., Гилхус Н.Е., Белен А., де Виссер М., Нолле Ф. Лечение постполиомиелитного синдрома. Кокрановская база данных Syst Rev . 2011 16 февраля. CD007818. [Медлайн].
Bertolasi L, Frasson E, Turri M, Gajofatto A, Bordignon M, Zanolin E. Рандомизированное контролируемое исследование внутривенного введения иммуноглобулина у пациентов с постполиомиелитным синдромом. J Neurol Sci . 2013 15 июля.330 (1-2): 94-9. [Медлайн].
Гонсалес Х., Хадеми М., Борг К., Олссон Т. Лечение постполиомиелитного синдрома внутривенным иммуноглобулином: устойчивые эффекты на качество жизни и экспрессию цитокинов после одного года наблюдения. J Нейровоспаление . 2012. 9: 167. [Медлайн].
Остлунд Г., Броман Л., Верхаген Л., Борг К. Лечение внутривенным иммуноглобулином у пациентов, перенесших полиомиелит: оценка ответивших. Дж. Neurol . 2012 декабрь 259 (12): 2571-8.[Медлайн].
Werhagen L, Borg K. Влияние внутривенного иммуноглобулина на боль у пациентов с постполиомиелитным синдромом. J Rehabil Med . 2011 ноябрь 43 (11): 1038-40. [Медлайн].
Лу Ф, Ван Х, Цзян Дж, Чен В., Ма Х, Ма Х. Эффективность процедур декомпрессии и слияния передней шейки матки для лечения мономерной амиотрофии: проспективное рандомизированное контролируемое исследование: клиническая статья. J Нейрохирургия позвоночника . 2013 Октябрь.19 (4): 412-9. [Медлайн].
Мулинье А., Мулонге А., Пиалу Г., Розенбаум В. Обратимое БАС-подобное заболевание при ВИЧ-инфекции. Неврология . 2001 25 сентября. 57 (6): 995-1001. [Медлайн].
Jubelt B, Berger JR. В основе БАС лежит вирусное заболевание? Уроки пандемии СПИДа. Неврология . 2001 25 сентября. 57 (6): 945-6. [Медлайн].
Кидд Д., Уильямс А.Дж., Ховард Р.С. Полиомиелит. Postgrad Med J .1996 ноябрь 72 (853): 641-7. [Медлайн]. [Полный текст].
Мышечная дистрофия — Симптомы и причины
Обзор
Мышечная дистрофия — это группа заболеваний, вызывающих прогрессирующую слабость и потерю мышечной массы. При мышечной дистрофии аномальные гены (мутации) препятствуют выработке белков, необходимых для формирования здоровых мышц.
Есть много видов мышечной дистрофии. Симптомы самого распространенного разнообразия начинаются в детстве, в основном у мальчиков.Другие типы не появляются до взрослого возраста.
Нет лекарства от мышечной дистрофии. Но лекарства и терапия могут помочь справиться с симптомами и замедлить течение болезни.
Продукты и услуги
Показать больше товаров от Mayo ClinicСимптомы
Основным признаком мышечной дистрофии является прогрессирующая мышечная слабость. Специфические признаки и симптомы начинаются в разном возрасте и в разных группах мышц, в зависимости от типа мышечной дистрофии.
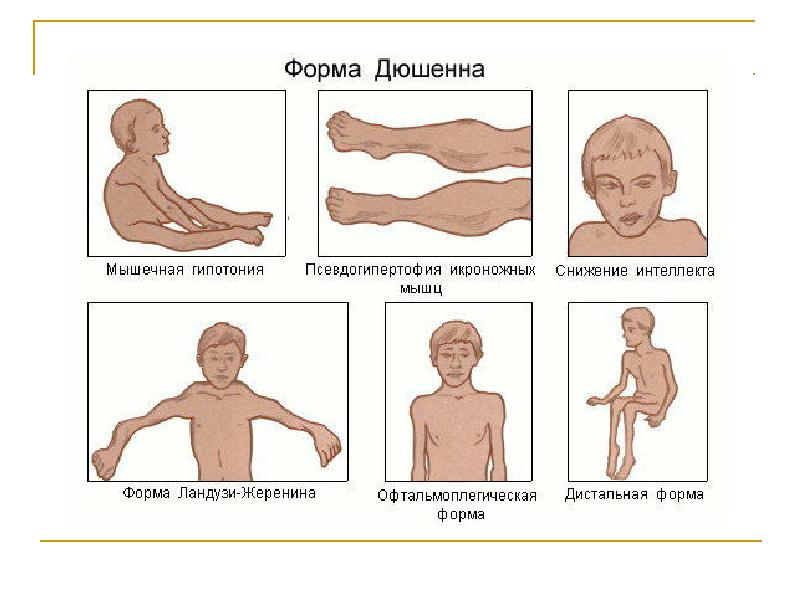
Мышечная дистрофия Дюшенна
Это наиболее распространенная форма. Хотя девочки могут быть переносчиками и иметь легкое поражение, это гораздо чаще встречается у мальчиков.
Признаки и симптомы, которые обычно появляются в раннем детстве, могут включать:
- Частые падения
- Затруднения при вставании из положения лежа или сидя
- Проблемы с бегом и прыжками
- Перевязывающая походка
- Ходьба на цыпочках
- Большие икроножные мышцы
- Мышечные боли и скованность
- Нарушения обучаемости
- Отсроченный рост
Мышечная дистрофия Беккера
Признаки и симптомы сходны с таковыми при мышечной дистрофии Дюшенна, но имеют тенденцию быть более легкими и прогрессировать медленнее.Симптомы обычно начинаются в подростковом возрасте, но могут появиться не ранее 20 лет или позже.
Другие типы мышечной дистрофии
Некоторые типы мышечной дистрофии определяются конкретным признаком или местом начала симптомов в организме. Примеры включают:
- Миотонический. Характеризуется неспособностью расслабить мышцы после сокращения. Обычно в первую очередь поражаются мышцы лица и шеи. Люди с этой формой обычно имеют длинные тонкие лица; опущенные веки; и лебединые шеи.
- Facioscapulohumeral (FSHD). Мышечная слабость обычно начинается с лица, бедер и плеч. Лопатки могут выступать, как крылья, когда руки подняты. Начало обычно происходит в подростковом возрасте, но может начаться в детстве или даже в возрасте 50 лет.
- Врожденный. Этот тип поражает мальчиков и девочек и проявляется при рождении или в возрасте до 2 лет. Некоторые формы прогрессируют медленно и вызывают лишь легкую инвалидность, в то время как другие быстро прогрессируют и вызывают серьезные нарушения.
- Конечностный пояс. Обычно в первую очередь поражаются мышцы бедра и плеча. Люди с этим типом мышечной дистрофии могут испытывать трудности с поднятием передней части стопы и часто спотыкаются. Начало обычно начинается в детстве или подростковом возрасте.
Когда обращаться к врачу
Обратитесь за медицинской помощью, если вы заметили признаки мышечной слабости — например, повышенную неуклюжесть и падение — у вас или вашего ребенка.
Причины
Определенные гены участвуют в производстве белков, защищающих мышечные волокна.Мышечная дистрофия возникает при дефекте одного из этих генов.
Каждая форма мышечной дистрофии вызывается генетической мутацией, характерной для данного типа заболевания. Большинство этих мутаций передаются по наследству.
Факторы риска
Мышечная дистрофия встречается у представителей обоих полов, всех возрастов и рас. Однако наиболее распространенная разновидность, Дюшенн, обычно встречается у мальчиков. Люди с семейным анамнезом мышечной дистрофии подвергаются более высокому риску развития заболевания или передачи его своим детям.
Осложнения
Осложнения прогрессирующей мышечной слабости включают:
- Проблемы при ходьбе. Некоторым людям с мышечной дистрофией со временем необходимо использовать инвалидное кресло.
- Проблемы с использованием оружия. Повседневная деятельность может стать труднее, если затронуты мышцы рук и плеч.
- Укорочение мышц или сухожилий вокруг суставов (контрактуры). Контракты могут еще больше ограничить мобильность.
- Проблемы с дыханием. Прогрессирующая слабость может влиять на мышцы, отвечающие за дыхание. Людям с мышечной дистрофией, возможно, в конечном итоге потребуется использовать устройство для искусственного дыхания (вентилятор), сначала ночью, но, возможно, также и в течение дня.
- Искривление позвоночника (сколиоз). Ослабленные мышцы могут не удерживать позвоночник прямо.
- Проблемы с сердцем. Мышечная дистрофия может снизить эффективность сердечной мышцы.
- Проблемы с глотанием. Если поражены мышцы, участвующие в процессе глотания, могут развиться проблемы с питанием и аспирационная пневмония. Питательные трубки могут быть вариантом.
31 января 2020 г.
| Тип | Возраст начала заболевания | Симптомы, скорость прогрессирования и ожидаемая продолжительность жизни |
|---|---|---|
| Беккер | от подросткового до раннего взросления |
Возраст в дебюте Возраст в дебюте Симптомы почти идентичны Дюшенну, но менее серьезны; прогрессирует медленнее, чем у Дюшена; дожить до среднего возраста.Как и в случае с Дюшенном, болезнь почти всегда ограничивается мужчинами. |
| Врожденный | рождение |
Возраст в дебюте Возраст в дебюте Симптомы включают общую мышечную слабость и возможные деформации суставов; болезнь медленно прогрессирует; сокращенная продолжительность жизни. |
| Дюшенна | От 2 до 6 лет |
Возраст в дебюте Возраст в дебюте Симптомы включают общую мышечную слабость и истощение; поражает таз, плечи и голени; в конечном итоге задействует все произвольные мышцы; выживание после 20 лет — редкость.Встречается только у мальчиков. Очень редко может повлиять на женщину, у которой гораздо более легкие симптомы и лучший прогноз. |
| Дистальный | От 40 до 60 лет |
Возраст в дебюте Возраст в дебюте Симптомы включают слабость и истощение мышц рук, предплечий и голеней; прогрессирование медленное; редко приводит к полной нетрудоспособности. |
| Эмери-Драйфус | от детства до раннего подросткового возраста |
Возраст в дебюте Возраст в дебюте Симптомы включают слабость и истощение мышц плеча, предплечья и голени; суставные деформации распространены; прогрессирование медленное; внезапная смерть может наступить из-за проблем с сердцем. |
| Лицево-лопаточно-плечевой | от детства к ранним взрослым |
Возраст в дебюте Возраст в дебюте Симптомы включают слабость и слабость лицевых мышц с некоторым истощением плеч и предплечий; прогрессирование медленное с периодами быстрого ухудшения; продолжительность жизни может составлять многие десятилетия после начала заболевания. |
| Ремень для конечностей | от позднего детства до среднего возраста |
Возраст в дебюте Возраст в дебюте Симптомы включают слабость и истощение, поражающие в первую очередь плечевой и тазовый пояс; прогрессирование медленное; смерть обычно наступает из-за сердечно-легочных осложнений. |
| Миотонический | От 20 до 40 лет |
Возраст в дебюте Возраст в дебюте Симптомы включают слабость всех групп мышц, сопровождающуюся замедленным расслаблением мышц после сокращения; поражает в первую очередь лицо, ступни, руки и шею; прогрессирование идет медленно, иногда от 50 до 60 лет. |
| Окулофарингеальный | От 40 до 70 лет |
Возраст в дебюте Возраст в дебюте Симптомы поражают мышцы век и горла, вызывая ослабление мышц горла, что со временем приводит к неспособности глотать и истощению из-за недостатка пищи; прогрессирование идет медленно. |
Спинальная мышечная атрофия (СМА) — Заболевания
Спинальная мышечная атрофия (СМА)
Что такое мышечная атрофия позвоночника?
Спинальная мышечная атрофия (СМА) — это генетическое заболевание, поражающее центральную нервную систему, периферическую нервную систему и произвольные движения мышц (скелетные мышцы).
Большинство нервных клеток, контролирующих мышцы, расположены в спинном мозге, что составляет слово spinal в названии болезни.SMA — это мышечной , потому что ее основное воздействие на мышцы, которые не получают сигналы от этих нервных клеток. Атрофия — медицинский термин, обозначающий уменьшение размеров, что обычно происходит с мышцами, когда они не стимулируются нервными клетками.
SMA включает потерю нервных клеток, называемых двигательных нейронов в спинном мозге, и классифицируется как болезнь двигательных нейронов.
В наиболее распространенной форме СМА (СМА 5 хромосомы или СМА, связанная с SMN) существует большой разброс по возрасту начала, симптомам и скорости прогрессирования.Чтобы учесть эти различия, связанная с 5-й хромосомой СМА, которая часто является аутосомно-рецессивной, классифицируется на типы с 1 по 4.
Возраст, в котором появляются симптомы СМА, примерно коррелирует со степенью нарушения двигательной функции: чем раньше возраст начала, тем сильнее влияние на двигательную функцию. Дети, у которых проявляются симптомы при рождении или в младенчестве, обычно имеют самый низкий уровень функционирования (тип 1). Позднее начало СМА с менее тяжелым течением (типы 2 и 3, а у подростков или взрослых — тип 4) обычно коррелирует со все более высокими уровнями двигательной функции.
Подробнее см. Формы SMA.
Что вызывает SMA?
Хромосома 5 SMA вызвана дефицитом белка мотонейрона, называемого SMN, для «выживания мотонейрона». Этот белок, как следует из его названия, по-видимому, необходим для нормальной функции двигательных нейронов. SMN играет ключевую роль в экспрессии генов в двигательных нейронах. Его дефицит вызван генетическими дефектами (мутациями) хромосомы 5 в гене SMN1 . Наиболее частая мутация в гене SMN1 у пациентов с диагнозом СМА — делеция целого сегмента, называемого экзоном 7. 1 Соседние гены SMN2 могут частично компенсировать нефункциональные гены SMN1, поскольку между этими двумя генами существует 99% идентичности. 2
Другие редкие формы SMA (не хромосомы 5) вызываются мутациями в генах, отличных от SMN1 . 3
Для получения дополнительной информации, включая подробные сведения о редкой, не связанной с хромосомой 5 СМА, см. «Формы СМА» и «Причины / наследование».
Каковы симптомы СМА?
Симптомы СМА охватывают широкий спектр, от легких до тяжелых.
Основным симптомом СМА, связанной с хромосомой 5 (SMN), является слабость произвольных мышц. Наиболее поражены мышцы, расположенные ближе всего к центру тела, например, плечи, бедра, бедра и верхняя часть спины. Кажется, что нижние конечности поражены больше, чем верхние, а глубокие сухожильные рефлексы снижены. 4
Особые осложнения возникают при поражении мышц, используемых для дыхания и глотания, что приводит к нарушению этих функций.Если мышцы спины слабеют, могут развиться искривления позвоночника.
Возраст начала и уровень двигательной функции, достигнутой при СМА, связанной с хромосомой 5, сильно различаются. Они примерно коррелируют с тем, сколько функционального белка SMN присутствует в мотонейронах, что, в свою очередь, коррелирует с количеством копий генов SMN2 , которые есть у человека. Сенсорные, умственные и эмоциональные функции при СМА 5-й хромосомы полностью нормальны.
Некоторые формы SMA не связаны с хромосомой 5 или дефицитом SMN.Эти формы сильно различаются по степени тяжести и наиболее пораженным мышцам. В то время как большинство форм, таких как форма, связанная с хромосомой 5, поражает в основном проксимальные мышцы, существуют другие формы, которые влияют в основном на дистальных мышц (те, что дальше от центра тела) — по крайней мере, вначале.
Для получения дополнительной информации см. Признаки и симптомы.
Как прогрессирует СМА?
При СМА, связанной с хромосомой 5, чем позже проявляются симптомы и чем больше в нем белка SMN, тем вероятнее будет более легкое течение заболевания.Если в прошлом младенцы со СМА обычно не выживали более двух лет, сегодня большинство врачей считают СМА, связанную с СМА, континуумом, и предпочитают не делать жестких прогнозов относительно продолжительности жизни или слабости, строго основанных на возрасте начала.
СМА — наиболее частая генетическая причина смертности младенцев.
Каков статус исследований SMA?
Исследования были сосредоточены на стратегиях увеличения производства в организме белка SMN, отсутствующего в формах заболевания, связанных с хромосомой 5.Подходы включают методы, помогающие двигательным нейронам выжить в неблагоприятных обстоятельствах.
23 декабря 2016 г. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило Spinraza (nusinersen) для лечения СМА. Спинраза предназначена для лечения основного дефекта СМА, что означает, что он потенциально может быть эффективным для замедления, остановки или, возможно, обращения симптомов СМА. Для получения дополнительной информации см. Спинраза одобрена.
В мае 2019 года FDA одобрило Zolgensma (онасемноген abeparvovac-xioi), первую генную заместительную терапию нервно-мышечного заболевания.Золгенсма — это одноразовое внутривенное (в вену) вливание для лечения детей младше 2 лет с СМА с биаллельными мутациями в гене SMN1 , включая тех, у которых на момент постановки диагноза отсутствуют симптомы. Для получения дополнительной информации прочтите, что FDA утверждает Zolgensma AveXis для лечения спинальной мышечной атрофии у педиатрических пациентов .
Для получения дополнительной информации см. Исследование SMA: впереди на полной скорости и в фокусе: мышечная атрофия позвоночника (SMA). Истории семей, живущих с SMA, можно найти в наших историях SMA на Strongly, в блоге MDA .
В августе 2020 года FDA одобрило рисдиплам (торговая марка Evrysdi *) для лечения СМА у взрослых и детей в возрасте двух месяцев и старше. Evysdi — это пероральный препарат, предназначенный для повышения уровня белка SMN за счет увеличения выработки «резервного» гена SMN2.
Загрузите наш информационный бюллетень о спинальной мышечной атрофии
Узнайте о реакции MDA на COVID-19
Список литературы
- Огино, С. и Уилсон, Р.Б. Генетическое тестирование и оценка риска спинальной мышечной атрофии (СМА). Human Genetics (2002). DOI: 10.1007 / s00439-002-0828-x
- Lefebvre, S. et al. Идентификация и характеристика гена, определяющего мышечную атрофию позвоночника. Ячейка (1995). DOI: 10.1016 / 0092-8674 (95)
-3
- Даррас, Б. Т. Спинальные мышечные атрофии, отличные от 5q: Буквенно-цифровой суп густеет. Неврология (2011). DOI: 10.1212 / WNL.0b013e3182267bd8
- Арнольд, В.Д., Кассар Д. и Киссель Дж. Т. Спинальная мышечная атрофия: диагностика и лечение в новую терапевтическую эру. Мышцы и нервы (2015). DOI: 10.1002 / mus.24497
Информационный бюллетень о спинальной мышечной атрофии
Что такое мышечная атрофия позвоночника?
Спинальная мышечная атрофия (СМА) — это группа наследственных заболеваний, которые постепенно разрушают двигательные нейроны — нервные клетки в стволе головного и спинного мозга, которые контролируют важную активность скелетных мышц, такую как речь, ходьба, дыхание и глотание, ведущие к мышечной слабости и атрофии.Моторные нейроны контролируют движения в руках, ногах, груди, лице, горле и языке. Когда возникают перебои в передаче сигналов между двигательными нейронами и мышцами, мышцы постепенно ослабевают, начинают истощаться и развиваются подергивания (так называемые фасцикуляции ) .
верх
Что вызывает СМА?
Наиболее распространенная форма СМА вызывается дефектами обеих копий гена выживания моторного нейрона 1 (SMN1) на хромосоме 5q. Этот ген производит белок выживания моторных нейронов (SMN), который поддерживает здоровье и нормальную функцию моторных нейронов.Люди с SMA имеют недостаточный уровень белка SMN, что приводит к потере мотонейронов в спинном мозге, вызывая слабость и истощение скелетных мышц. Эта слабость часто более выражена в мышцах туловища, верхней части ног и рук, чем в мышцах рук и ног.
Существует много типов мышечной атрофии позвоночника, которые вызваны изменениями одних и тех же генов. Менее распространенные формы SMA вызываются мутациями в других генах, включая ген VAPB, расположенный на хромосоме 20, ген DYNC1h2 на хромосоме 14, ген BICD2 на хромосоме 9 и ген UBA1 на X-хромосоме.Типы различаются по возрасту начала и степени мышечной слабости; однако типы пересекаются.
верх
Как передается по наследству?
За исключением редких случаев, вызванных мутациями в гене UBA1, СМА наследуется по аутосомно-рецессивному типу , что означает, что пораженный человек имеет два мутировавших гена, часто наследуя по одному от каждого родителя. Те, кто несет только один мутировавший ген, являются носителями болезни без каких-либо симптомов.Аутосомно-рецессивные заболевания могут поражать более одного человека в одном поколении (братьев и сестер или двоюродных братьев и сестер).
верх
Какие бывают типы SMA?
Существует широкий спектр нарушений, наблюдаемых при СМА, вызванных дефектами гена SMN1, от начала до рождения с затрудненным дыханием при рождении до легкой слабости у взрослых. Соответственно, эту наиболее распространенную форму СМА можно разделить на четыре типа в зависимости от достигнутого наивысшего двигательного рубежа.
- СМА типа I, также называемая болезнью Верднига-Гофмана или СМА с младенческим началом, обычно проявляется в возрасте до 6 месяцев.Младенцы с наиболее тяжелым поражением (тип СМА 0 или IA) имеют ограниченные движения даже в утробе матери, рождаются с контрактурами и затрудненным дыханием, причем смерть наступает в первый год жизни без лечения. Симптомы СМА типа I включают гипотонию (снижение мышечного тонуса), снижение движений конечностей, отсутствие сухожильных рефлексов, фасцикуляции, затруднения глотания и кормления, а также нарушение дыхания. У этих детей также с возрастом развивается сколиоз (искривление позвоночника) или другие аномалии скелета.Без какого-либо лечения пораженные дети никогда не сидят и не стоят, и подавляющее большинство обычно умирает от дыхательной недостаточности в возрасте до 2 лет. Дети с СМА типа I теперь живут дольше и могут достичь более высоких двигательных навыков, таких как сидение и даже ходьба, благодаря более активной клинической помощи и недавно доступному лечению, изменяющему заболевание.
- Дети с СМА типа II, промежуточной формой , обычно проявляют первые симптомы в возрасте от 6 до 18 месяцев, хотя некоторые могут проявиться раньше.Они могут сидеть без поддержки, но не могут стоять или ходить без посторонней помощи, а некоторые могут потерять способность самостоятельно сидеть без помощи со временем. У них могут быть проблемы с дыханием, включая гиповентиляцию во сне. Без лечения болезнь прогрессирует. Ожидаемая продолжительность жизни сокращается, но большинство людей доживают до подросткового или молодого возраста. Благодаря лечению, изменяющему болезнь, и проактивной клинической помощи, у детей со СМА типа II улучшились двигательные исходы.
- Дети с СМА типа III (болезнь Кугельберга-Веландера) развивают симптомы после 18 месяцев и могут самостоятельно ходить. Сначала они испытывают трудности при ходьбе и беге, подъеме по ступенькам или вставании со стула. Чаще всего первыми поражаются проксимальные мышцы ног, при этом наблюдается тремор в руках. Осложнения включают сколиоз и контрактуры суставов — хроническое укорочение мышц или сухожилий вокруг суставов — вызванные ненормальным мышечным тонусом и слабостью, которые препятствуют свободному движению суставов.Люди со СМА типа III могут быть предрасположены к респираторным инфекциям, но при осторожном уходе у большинства из них продолжительность жизни нормальная. Лечение, изменяющее заболевание, может улучшить результаты.
- У лиц со СМА типа IV симптомы заболевания развиваются после 21 года, включая слабость проксимальных мышц от легкой до умеренной степени, а также другие симптомы.
верх
Как диагностируется СМА?
Доступен анализ крови для поиска делеций или мутаций гена SMN1. Этот тест выявляет не менее 95 процентов СМА типов I, II и III, а также может выявить, является ли человек носителем дефектного гена, который может передаваться детям.Если не обнаружено отклонений в гене SMN1 или если история пациента и обследование не типичны для СМА, другие диагностические тесты могут включать электромиографию (которая регистрирует электрическую активность мышц во время сокращения и в состоянии покоя), исследования скорости нервной проводимости (которые измерение способности нерва посылать электрический сигнал), биопсия мышц (используется для диагностики многих нервно-мышечных расстройств) и другие анализы крови.
верх
Существуют ли методы лечения СМА?
Полного излечения SMA не существует.Лечение заключается в устранении симптомов и предотвращении осложнений.
В декабре 2016 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобрило нусинерсен (Спинраза ™) в качестве первого препарата, одобренного для лечения детей и взрослых со СМА. Препарат вводят путем инъекции в жидкость, окружающую спинной мозг. Он разработан для увеличения производства полноразмерного белка SMN, который имеет решающее значение для поддержания двигательных нейронов. Преимущество лучше всего документировано у младенцев и детей, особенно при раннем начале лечения.Несколько других методов лечения находятся на поздних стадиях разработки и могут стать доступными для больных в ближайшем будущем.
В мае 2019 года FDA одобрило генную терапию онасемногеном abeparovec-xioi (Zolgensma ™) для детей младше 2 лет, у которых СМА младенческого начала. Безопасный вирус доставляет полнофункциональный ген SMN человека к целевым двигательным нейронам, что, в свою очередь, улучшает движение и функцию мышц, а также улучшает выживаемость. В августе 2020 года FDA одобрило пероральный препарат рисдиплам (Evrysdi) для лечения пациентов с СМА в возрасте двух месяцев и старше.
Физиотерапия, трудотерапия и реабилитация могут помочь улучшить осанку, предотвратить неподвижность суставов и замедлить мышечную слабость и атрофию. Упражнения на растяжку и укрепление могут помочь уменьшить контрактуры, увеличить диапазон движений и поддерживать циркуляцию крови. Некоторым людям требуется дополнительная терапия по поводу проблем с речью и глотанием. Вспомогательные устройства, такие как опоры или скобы, ортопедические стельки, синтезаторы речи и инвалидные коляски, могут быть полезны для улучшения функциональной независимости.
Правильное питание и калорийность необходимы для поддержания веса и силы, избегая при этом длительного голодания. Людям, которые не могут жевать или глотать, может потребоваться введение зонда для кормления. Неинвазивная вентиляция в ночное время может улучшить дыхание во время сна, а некоторым людям также может потребоваться вспомогательная вентиляция в течение дня из-за мышечной слабости в шее, горле и груди.
верх
Каков прогноз?
Прогноз зависит от типа SMA.Некоторые формы СМА без лечения приводят к летальному исходу.
Люди со СМА могут казаться стабильными в течение длительного времени, но не следует ожидать улучшения без лечения.
верх
Какие исследования проводятся?
Национальный институт неврологических расстройств и инсульта (NINDS), входящий в состав Национальных институтов здравоохранения (NIH), проводит фундаментальные, трансляционные и клинические исследования СМА в лабораториях Национального института здоровья, а также поддерживает исследования посредством грантов крупным медицинским учреждениям. через всю страну.
Клеточные и молекулярные исследования стремятся понять механизмы, которые запускают дегенерацию мотонейронов.
Ученые проанализировали ткани человека и разработали широкий спектр модельных систем на животных и клетках для исследования процессов болезни и ускорения тестирования потенциальных методов лечения. Среди этих усилий:
- Было показано, что генная терапия и специфические препараты останавливают разрушение двигательных нейронов и замедляют прогрессирование заболевания на моделях мышей и лиц с СМА.NINDS поддержал исследования по разработке этих методов и предоставлению возможностей для клинических испытаний на пациентах. Клинические испытания генной терапии при СМА продолжаются.
- Животные модели СМА представляют собой важные инструменты в открытии и разработке новых методов лечения СМА. Ученые разработали модели рыбок данио, мышей и свиней, включая модели менее тяжелых типов СМА 2 и 3, которые могут значительно помочь в идентификации новых терапевтических мишеней и возможных методов лечения.
Ученые, поддерживаемые Национальным институтом здравоохранения (NIH), собрали данные о детях с предсимптоматическими или недавно диагностированными заболеваниями со СМА типа 1, 2 или 3 и их здоровых братьях и сестрах.Цель этого исследования — предоставить родителям консультации и разъяснить возможные возможности проведения клинических испытаний.
NINDS учредила сеть клинических испытаний NeuroNext (Сеть передового опыта в области нейробиологии NINDS), чтобы способствовать быстрой разработке и внедрению исследований по неврологическим расстройствам, поражающим взрослых и / или детей. Среди своих целей сеть предназначена для разработки ранней фазы испытаний, направленных на выявление биомаркеров — обычно физических признаков или веществ в крови или других жидкостях организма, которые могут быть измерены для определения наличия и тяжести заболевания — и многообещающих испытаний. новые методы лечения.Один из проектов заключался в определении биомаркеров СМА и понимании причины и механизмов, лежащих в основе заболевания. Данные естественной истории, полученные в ходе этого исследования, привели к одобрению nusinersen. Знания, полученные в результате этого исследования, улучшили дизайн дополнительных клинических испытаний СМА.
Где я могу получить дополнительную информацию?
Для получения дополнительной информации о неврологических расстройствах или исследовательских программах, финансируемых Национальным институтом неврологических расстройств и инсульта, обращайтесь в Сеть мозговых ресурсов и информации Института (BRAIN) по телефону:
МОЗГ
стр.О. Box 5801
Bethesda, MD 20824
800-352-9424
Информацию также можно получить в следующих организациях:
Cure SMA
925 Busse Road
Elk Grove Village, IL 60007
847-367-7620
800-886-1762
Фонд спинальной мышечной атрофии
126 East 56th Street
30-й этаж
New York, NY 10022
646-253-7100
877-386-3762
Ассоциация мышечной дистрофии
161 N.Clarke,
Suite 3550
Chicago, IL 60601
800-572-1717
«Информационный бюллетень о спинальной мышечной атрофии», NINDS, дата публикации май 2019 г.
Публикация NIH № 19-NS-5597
Вернуться на страницу с информацией о спинальной мышечной атрофии
См. Список всех расстройств NINDS
Publicaciones en Español
Атрофия мышечно-спинномозговая
Подготовлено:
Офис по связям с общественностью
Национальный институт неврологических расстройств и инсульта
Национальные институты здравоохранения
Bethesda, MD 20892
NINDS, связанные со здоровьем, предоставляются только в информационных целях и не обязательно представляют собой поддержку или официальную позицию Национального института неврологических расстройств и инсульта или любого другого федерального агентства.Консультации по лечению или уходу за отдельным пациентом следует получать после консультации с врачом, который обследовал этого пациента или знаком с историей болезни этого пациента.
Вся информация, подготовленная NINDS, является общественным достоянием и может свободно копироваться. Благодарность NINDS или NIH приветствуется.
Спинальная мышечная атрофия (СМА): типы, симптомы и лечение
Обзор
Что такое спинальная мышечная атрофия (СМА)?
Спинальная мышечная атрофия (СМА) — это генетическое (наследственное) нервно-мышечное заболевание, из-за которого мышцы становятся слабыми и истощаются.Люди с СМА теряют нервные клетки определенного типа в спинном мозге (называемые мотонейронами), которые контролируют движение мышц. Без этих мотонейронов мышцы не получают нервных сигналов, которые заставляют мышцы двигаться. Слово атрофия — это медицинский термин, который означает меньшее. При СМА некоторые мышцы становятся меньше и слабее из-за недостаточной активности.
Насколько распространена мышечная атрофия позвоночника?
Приблизительно от 10 000 до 25 000 детей и взрослых живут с СМА в Соединенных Штатах. Это редкое заболевание, которым страдает один из 6000–10 000 детей.
Кто может получить мышечную атрофию позвоночника?
Человек с СМА наследует две копии отсутствующего или дефектного (мутировавшего) гена моторного нейрона выживания 1 (SMN1). Один дефектный ген происходит от матери, а другой — от отца. Взрослый человек может иметь единственную копию дефектного гена, вызывающего СМА, и не знать об этом.
Около шести миллионов американцев (1 из 50) являются носителями мутированного гена SMN1. Эти носители имеют один здоровый ген SMN1 и один отсутствующий или дефектный ген SMN1. У перевозчиков не развивается SMA.Вероятность того, что у двух носителей будет ребенок с СМА, составляет 1 из 4.
Какие бывают типы мышечной атрофии позвоночника?
Существует четыре основных типа SMA:
- Тип 1 (тяжелый): Около 60% людей со СМА имеют тип 1, также называемый болезнью Верднига-Хоффмана. Симптомы появляются при рождении или в течение первых шести месяцев жизни младенца. Младенцы с СМА типа 1 испытывают трудности с глотанием и сосанием. Они не справляются с обычными вехами, такими как поднятие головы или сидение.По мере того как мышцы продолжают ослабевать, дети становятся более склонными к респираторным инфекциям и коллапсу легких (пневмотораксу). Большинство детей с СМА типа 1 умирают до своего второго дня рождения.
- Тип 2 (промежуточный): Симптомы СМА 2 типа (также называемой болезнью Дубовица) появляются, когда ребенку от шести до 18 месяцев. Этот тип чаще поражает нижние конечности. Дети со СМА 2 типа могут сидеть, но не могут ходить. Большинство детей со СМА 2 типа доживают до взрослого возраста.
- Тип 3 (легкая форма): Симптомы СМА 3 типа (также называемой СМА Кугельберта-Веландера или СМА с ювенильным началом) появляются после первых 18 месяцев жизни ребенка. У некоторых людей с типом 3 симптомы болезни проявляются только в раннем взрослом возрасте. Симптомы 3-го типа включают легкую мышечную слабость, затруднения при ходьбе и частые респираторные инфекции. Со временем симптомы могут повлиять на способность ходить или стоять. СМА типа 3 не приводит к значительному сокращению продолжительности жизни.
- Тип 4 (взрослый): Редкая взрослая форма СМА обычно не проявляется до середины 30-х годов.Симптомы мышечной слабости прогрессируют медленно, поэтому большинство людей с типом 4 остаются подвижными и живут полноценной жизнью.
Симптомы и причины
Что вызывает мышечную атрофию позвоночника?
У людей с СМА отсутствует часть гена SMN1 или изменен (мутирован) ген. Здоровый ген SMN1 производит белок SMN. Моторным нейронам нужен этот белок, чтобы выжить и нормально функционировать.
Люди с СМА не вырабатывают достаточное количество белка SMN, поэтому мотонейроны сжимаются и умирают.В результате мозг не может контролировать произвольные движения, особенно движения в голове, шее, руках и ногах.
У людей также есть гены SMN2, которые производят небольшое количество белка SMN. У человека может быть до восьми копий гена SMN2. Наличие нескольких копий гена SMN2 обычно приводит к менее серьезным симптомам СМА, поскольку дополнительные гены восполняют недостающий белок SMN1. В редких случаях СМА вызывают мутации гена, не относящегося к SMN (не хромосомы 5).
Каковы симптомы мышечной атрофии позвоночника?
Симптомы СМАразличаются в зависимости от типа.В целом, люди с СМА испытывают прогрессирующую потерю мышечного контроля, движения и силы. Потеря мышечной массы ухудшается с возрастом. Заболевание имеет тенденцию серьезно поражать мышцы, расположенные ближе всего к туловищу и шее. Некоторые люди с СМА никогда не ходят, не сидят и не стоят. Другие постепенно теряют способность выполнять эти действия.
Диагностика и тесты
Как диагностируется мышечная атрофия позвоночника?
Некоторые симптомы СМА напоминают симптомы нервно-мышечных расстройств, таких как мышечная дистрофия.Чтобы определить причину симптомов, ваш лечащий врач проведет медицинский осмотр и получит историю болезни. Ваш врач может также назначить один или несколько из этих тестов для диагностики SMA:
- Анализ крови: Анализ крови на ферменты и белки позволяет проверить высокий уровень креатинкиназы. Ухудшенные мышцы высвобождают этот фермент в кровоток.
- Генетический тест: Этот анализ крови выявляет проблемы с геном SMN1. В качестве диагностического инструмента генетический тест на 95% эффективен при обнаружении измененного гена SMN1.В некоторых штатах тест на СМА проводится в рамках плановых обследований новорожденных.
- Тест нервной проводимости: Электромиограмма (ЭМГ) измеряет электрическую активность нервов, мышц и нервов.
- Биопсия мышцы: В редких случаях врач может выполнить биопсию мышцы. Эта процедура включает удаление небольшого количества мышечной ткани и отправку ее в лабораторию для исследования. Биопсия может показать атрофию или потерю мышечной массы.
Можно ли диагностировать мышечную атрофию позвоночника во время беременности?
Если вы беременны и имеете семейный анамнез СМА, пренатальные тесты могут определить, есть ли это заболевание у вашего будущего ребенка.Эти тесты немного повышают риск выкидыша или прерывания беременности. Пренатальные тесты на SMA включают:
- Амниоцентез: Во время амниоцентеза ваш акушер вводит вам в живот тонкую иглу, чтобы извлечь небольшое количество жидкости из амниотического мешка. Лабораторный специалист (патолог) проверяет жидкость на наличие СМА. Этот тест проводится после 14 недели беременности.
- Взятие пробы ворсинок хориона (CVS): Ваш акушер берет небольшой образец ткани из плаценты через шейку матки или желудок.Патологоанатом проверяет образец на SMA. CVS может иметь место уже на 10 неделе беременности.
Ведение и лечение
Как справляется или лечится спинальная мышечная атрофия?
Нет лекарства от SMA. Лечение зависит от типа и симптомов СМА. Многие люди с СМА получают пользу от физиотерапии и эрготерапии и вспомогательных устройств, таких как ортопедические скобы, костыли, ходунки и инвалидные коляски.
Эти методы лечения также могут помочь:
- Терапия, модифицирующая заболевание: Эти препараты стимулируют выработку белка SMN.Нусинерсен (Спинраза®) предназначен для детей в возрасте от 2 до 12 лет. Ваш врач вводит препарат в пространство вокруг позвоночного канала. Другой препарат, рисдаплам (Эврисди®), помогает взрослым и детям старше двух месяцев. Люди принимают рисдаплам ежедневно внутрь (перорально).
- Заместительная генная терапия: Детям младше двух лет может быть полезна однократная внутривенная (IV) инфузия препарата, называемого онасемногеном абепарвовец-xioi (Zolgensma®). Эта терапия заменяет отсутствующий или неисправный ген SMN1 функционирующим геном.
Каковы осложнения мышечной атрофии позвоночника?
Со временем люди с СМА испытывают прогрессирующую мышечную слабость и потерю мышечного контроля. Возможные осложнения включают:
Профилактика
Как предотвратить мышечную атрофию позвоночника?
СМА — наследственное заболевание. Если вы или ваш партнер несете мутировавший ген, вызывающий СМА, генетический консультант может объяснить шансы, что ваш ребенок болен СМА или является носителем.
Возможно, вы сможете принять меры до беременности, чтобы снизить риск передачи СМА. Процесс, называемый предимплантационной генетической диагностикой (ПГД), позволяет идентифицировать эмбрионы, у которых нет мутировавшего гена. Ваш врач имплантирует здоровые эмбрионы во время экстракорпорального оплодотворения (ЭКО). PGD гарантирует, что ваш ребенок будет иметь два здоровых гена SMN1 и не заболеет SMA.
Перспективы / Прогноз
Каков прогноз (перспективы) для людей со спинальной мышечной атрофией?
Качество и продолжительность жизни людей с СМА варьируются в зависимости от типа.Младенцы со СМА типа 1 обычно умирают до своего второго дня рождения. Дети с СМА 2 или 3 типа могут жить полноценной жизнью в зависимости от тяжести симптомов. Люди, у которых СМА развивается в зрелом возрасте (тип 4), часто остаются активными и имеют нормальную продолжительность жизни.
Жить с
Когда мне позвонить врачу?
Вы должны позвонить своему врачу, если у кого-то есть SMA:
Какие вопросы я должен задать своему врачу?
Вы можете спросить:
- Как я или мой ребенок заболели СМА?
- Какой тип SMA есть у меня или моего ребенка?
- Каков прогноз для этого типа SMA?
- Как лучше всего лечить этот тип СМА?
- Каковы риски лечения и побочные эффекты?
- Подвержены ли другие члены семьи риску заболеть СМА? Если да, должны ли мы сдавать генетические тесты?
- Какой вид постоянного ухода понадобится мне или моему ребенку?
- Стоит ли обращать внимание на признаки осложнений?
Записка из клиники Кливленда
SMA — это генетическое нервно-мышечное заболевание, которое может существенно повлиять на качество и продолжительность жизни.Это прогрессирующее заболевание, которое со временем ухудшается. Симптомы могут присутствовать при рождении (тип 1), развиваться в детстве (тип 2 или 3) или во взрослом возрасте (тип 4). Многообещающие новые методы лечения заболеваний и замены генов. Можно нести ген, вызывающий СМА, и не знать об этом. Если СМА присутствует в вашей семье, поговорите со своим врачом о том, как снизить вероятность развития СМА у вашего будущего ребенка.
Мышечная дистрофия против мышечной атрофии: симптомы и многое другое
Атрофия мышц относится к сокращению или «истощению» мышц.Обычно это симптом другого состояния, а не состояние само по себе. Помимо потери размера мышц, мышечная атрофия также может вызвать мышечную слабость.
Мышечная дистрофия — это редкое генетическое заболевание, которое влияет на белки, которые создают и поддерживают здоровые мышцы. Хотя мышечная дистрофия может вызывать атрофию мышц, это разные состояния с разными причинами, симптомами и методами лечения.
SolStock / Getty Images
Причины
Причины мышечной дистрофии-
Спонтанная мутация гена
-
Унаследованная мутация аутосомно-доминантного гена
-
Наследственная мутация аутосомно-рецессивного гена
-
Унаследованная мутация Х-сцепленного гена
Причины мышечной дистрофии
Все типы мышечной дистрофии вызваны генетической мутацией, но не все случаи мышечной дистрофии передаются по наследству.
Не наследуются
Спонтанные мутации могут происходить в одном из тысяч генов, которые программируют белки, необходимые для создания и поддержания мышц. Это может привести к изменению, недостаточному количеству или отсутствию белков и нарушению нормальной работы клеток организма.
Хотя эти спонтанные мутации не наследуются от родителей, они могут передаваться детям человека с мышечной дистрофией, что приводит к наследственной мышечной дистрофии, или человеку, который является носителем мутировавшего гена, который может вызывать мышечную дистрофию.
Унаследовано
Человек получает 23 хромосомы от каждого родителя — одну половую хромосому и 22 неполовые хромосомы — всего 46 хромосом, расположенных парами.
Каждая из этих пар хромосом содержит две одинаковые хромосомы, за исключением половых хромосом. Люди, несущие две Х-хромосомы (женский генетический пол), передают одну из своих двух Х-хромосом своему потомству. Люди, несущие хромосомы XY (мужской генетический пол), передадут своим потомкам либо X, либо Y-хромосому.
Мутировавшие гены могут передаваться потомству через любую из этих хромосом от любого из родителей.
Мутировавший ген, который может вызвать мышечную дистрофию, может быть унаследован одним из трех способов:
- Аутосомно-доминантный: Мутировавший ген встречается на любой из неполовых хромосом, и только один родитель должен передать дефектный ген, чтобы вызвать заболевание. Это заболевание разовьется, если потомство получит дефектный ген от одного родителя или дефектный ген от каждого родителя.В этом случае нет перевозчиков. Если у человека дефектный ген, у него заболевание.
- Аутосомно-рецессивный: Чтобы заболевание проявилось, у человека должны быть два дефектных гена (по одному от каждого родителя). Родители не обязательно должны иметь это заболевание, они могут быть только носителями, то есть у каждого из них есть только один дефектный ген и один нормальный ген. Если у двух человек-носителей есть дети, у ребенка есть 25% -ный шанс получить оба дефектных гена и иметь заболевание, 25% -ный шанс того, что ребенок не получит дефектных генов, не будет иметь расстройства и не будет носителем, и 50-процентный шанс. % вероятность того, что ребенок получит только один дефектный ген и станет носителем.
- Х-сцеплено (сцеплено с полом): Это связано с мутацией гена на одной из половых хромосом. При мышечной дистрофии поражается Х-хромосома. Родитель с двумя X-хромосомами может передать либо затронутую X-хромосому (если у них есть хотя бы одна пораженная X), либо незатронутая X (если они являются только носителем). Родители с XY-хромосомами могут передавать мутировавший ген только в том случае, если у них есть заболевание, и только если они передают X, а не Y-хромосому.
Наследственная мышечная дистрофия и дети
Вот пример того, как это отображается:
- Ребенок с пораженной Х-хромосомой и Y-хромосомой будет иметь заболевание.
- Ребенок с одним пораженным X и одним здоровым X будет носителем и вряд ли проявит симптомы.
- Ребенок с двумя пораженными Х-хромосомами будет иметь заболевание, но в случае мышечной дистрофии это очень редко.
Причины атрофии мышц
Поскольку атрофия мышц — это в первую очередь симптом, она может быть вызвана рядом факторов.
Они могут включать:
- Травма
- Недоедание
- Болезнь
- Отсутствие активности, например, постельный режим
- Повреждение нерва
- Старение
- Бернс
- Другие состояния здоровья (как генетические, так и приобретенные), влияющие на мышечную систему, такие как мышечная дистрофия, остеоартрит и ревматоидный артрит
Типы
Типы мышечной дистрофии-
Мышечная дистрофия Дюшенна
-
Мышечная дистрофия Беккера
-
Врожденная мышечная дистрофия
-
Дистальная мышечная дистрофия
-
Мышечная дистрофия Эмери-Дрейфуса
-
Лице-лопаточно-плечевая мышечная дистрофия
-
Конечностно-поясная мышечная дистрофия
-
Миотоническая мышечная дистрофия
-
Окулофарингеальная мышечная дистрофия
-
Физиологические (неиспользованные)
-
Патологическое
-
Нейрогенный
Типы мышечной дистрофии
Хотя мышечная дистрофия относится к более чем 30 генетическим заболеваниям, существует девять основных типов.
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна — это форма мышечной дистрофии, которая имеет тенденцию быстро ухудшаться. Дополнительная информация о мышечной дистрофии Дюшенна включает:
- Самый распространенный из всех типов мышечной дистрофии
- Результат отсутствия протеина дистрофина (протеина, обнаруженного в мышцах, который помогает мышцам оставаться здоровыми и сильными)
- Х-сцеплено (мутированный ген находится на Х-хромосоме)
- В основном поражает людей с XY-хромосомами (две пораженные X-хромосомы встречаются редко)
- Носители (люди с одним пораженным Х и одним здоровым Х) могут проявлять некоторые симптомы, но если симптомы появляются, они обычно более легкие.
- Обычно проявляется в детстве
- Поражает около шести из каждых 100 000 детей в Северной Америке и Европе
- Вызывает прогрессирующую слабость и мышечную атрофию
- Сначала поражается верхняя часть ног и предплечья
- В конечном итоге влияет на все произвольные мышцы
- Может вызывать другие проблемы со здоровьем легких, сердца, позвоночника и других областей
- Без агрессивного лечения ожидаемая продолжительность жизни от позднего подросткового возраста до начала 20-х годов
- Современные методы лечения улучшили качество и продолжительность жизни (некоторые люди с мышечной дистрофией Дюшенна доживают до 30-40 лет)
Мышечная дистрофия Беккера
Мышечная дистрофия Беккера — вторая по распространенности мышечная дистрофия.Дополнительная информация о мышечной дистрофии Беккера включает:
- В основном поражает людей с хромосомами XY
- Приблизительно у одного из 18 000–30 000 американцев с XY-хромосомами развивается мышечная дистрофия Беккера
- Возраст появления симптомов обычно приходится на подростковый возраст, но может возникнуть в любом возрасте от 5 до 60 лет
- Вызвано мутациями в том же гене, что и мышечная дистрофия Дюшенна
- Подобно мышечной дистрофии Дюшенна, но обычно менее выражено
- Частичная, но недостаточная функция белка дистрофина
- Скорость прогрессирования мышечной слабости и атрофии сильно различается у разных людей
- Ожидаемая продолжительность жизни, как правило, средний возраст и старше
- Сначала поражается верхняя часть ног и предплечья
- Сердечные осложнения при мышечной дистрофии Беккера менее последовательны, чем при мышечной дистрофии Дюшенна, но в некоторых случаях могут быть столь же серьезными
- Когнитивные и поведенческие нарушения могут возникать, но не столь часты или серьезны, как при мышечной дистрофии Дюшенна
Врожденная мышечная дистрофия
Врожденная мышечная дистрофия — это наследственное заболевание, которое преимущественно поражает мышцы, мозг и глаза.Дополнительная информация включает:
- Группа аутосомно-рецессивных мышечных дистрофий, которые присутствуют при рождении или проявляются в возрасте до двух лет
- В равной степени влияет на всех полов
- Дегенерация мышц может быть легкой или тяжелой и в первую очередь поражает скелетные мышцы
- Тяжесть и прогрессирование мышечной слабости и дегенерации различаются в зависимости от типа заболевания
- Дефекты протеина мерозина являются причиной почти половины случаев врожденной мышечной дистрофии
- Встречается примерно у одного из 100000 человек всех возрастов
Дистальная мышечная дистрофия
Дистальная мышечная дистрофия (ДД) — это группа редких заболеваний, поражающих ваши мышцы (генетические миопатии).Дополнительная информация включает:
- Также называется дистальной миопатией
- Группа по крайней мере из шести специфических мышечных заболеваний, которые в первую очередь поражают дистальные мышцы (предплечья, кисти, голени и ступни)
- Поражает менее одного человека из 100000
- В равной степени влияет на всех полов
- Начало обычно в возрасте от 40 до 60 лет
- Обычно менее тяжелая, прогрессирует медленнее и затрагивает меньшее количество мышц, чем другие формы мышечной дистрофии
- Может распространяться на другие мышцы позже по мере прогрессирования болезни
- Может поражать сердце и дыхательные мышцы
- Преимущественно аутосомно-доминантное заболевание, но у молодых людей были зарегистрированы аутосомно-рецессивные формы
Мышечная дистрофия Эмери-Дрейфуса
Мышечная дистрофия Эмери-Дрейфуса — это заболевание, которое в первую очередь поражает мышцы, используемые для движения (скелетные мышцы) и сердце (сердечная мышца).Дополнительная информация включает:
- Может поражать горло, плечи и бедра
- Имеет две формы: рецессивную с Х-хромосомой и аутосомно-доминантную
- В первую очередь поражает людей с хромосомами XY
- Симптомы обычно проявляются к 10 годам, но могут появиться и к середине 20-х
- Сердечные симптомы могут быть самыми ранними и наиболее серьезными симптомами, иногда возникающими до появления мышечной слабости
- Люди с ХХ-хромосомами, являющиеся носителями, могут испытывать сердечные осложнения без мышечной слабости
- Часто со смертельным исходом в среднем возрасте из-за прогрессирующей легочной или сердечной недостаточности
Facioscapulohumeral Muscular Dystrophy
Лицо-лопаточно-плечевая мышечная дистрофия получила свое название от мышц, которые поражаются чаще всего: мышц лица, вокруг лопаток и плеч.Дополнительная информация включает:
- Также известна как болезнь Ландузи-Дежерина
- Третья по распространенности форма мышечной дистрофии, которой страдают около четырех человек из 100 000 в США.
- В равной степени влияет на всех полов
- Аутосомно-доминантное заболевание
- Может поражать глаза, уши и голени
- Обычно начинается в подростковом возрасте, но симптомы могут проявляться уже в детстве или даже в возрасте 40 лет
- Люди с этой формой мышечной дистрофии обычно имеют нормальную продолжительность жизни, но некоторые становятся инвалидами
- Мышечная слабость обычно асимметричная
- Обычно медленное прогрессирование с периодическими скачками быстрого разрушения мышц
Конечностно-поясная мышечная дистрофия
Мышечная дистрофия конечностей — это термин, обозначающий группу заболеваний, которые вызывают слабость и истощение мышц рук и ног.Дополнительная информация включает:
- Группа из более чем 20 наследственных состояний, включающих прогрессирующую потерю мышечной массы и симметричное ослабление произвольных мышц, ближайших к телу (особенно в плечах и вокруг бедер)
- Сердце, позвоночник, бедра, икры и туловище также могут быть поражены
- Поражает примерно двух человек из 100 000 в США.
- В равной степени влияет на всех полов
- Аутосомно-рецессивные типы мышечной дистрофии конечностей и пояса встречаются чаще, чем доминирующие формы, и обычно начинаются в детстве или подростковом возрасте
- Аутосомно-доминантные типы мышечной дистрофии конечностей и пояса обычно появляются в зрелом возрасте
- Скорость прогрессирования, тяжесть и последовательность прогрессирования различаются в зависимости от типа, но, как правило, чем раньше появляются симптомы, тем быстрее скорость прогрессирования заболевания
- Многие люди с мышечной дистрофией конечностей становятся инвалидами в течение 20 лет после начала заболевания
Миотоническая мышечная дистрофия
Миотоническая дистрофия — наиболее распространенная форма мышечной дистрофии, которая начинается в зрелом возрасте.Дополнительная информация включает:
- Также известна как болезнь Штейнерта и миотоническая дистрофия
- Поражает взрослых европейцев
- Поражает примерно 10 человек из 100 000
- Единственная форма мышечной дистрофии, которая проявляется миотонией (неспособность расслабить мышцы после внезапного сокращения), но миотония также встречается при других недистрофических мышечных заболеваниях
- Типичное начало — в возрасте от 20 до 30 лет, но может произойти где угодно в диапазоне от рождения до 70 лет
- Может поражать многие части тела, включая сначала лицо, шею, руки, кисти, бедра и голени, затем сердце, легкие, желудок, кишечник, мозг, глаза и органы, вырабатывающие гормоны.
- Тяжелая форма миотонической мышечной дистрофии может возникнуть при рождении и почти исключительно у детей, унаследовавших дефектный ген от своего биологического родителя с хромосомами ХХ.У родителя может быть очень мало или легкие симптомы, и он может не осознавать, что у него заболевание, до тех пор, пока не родится больной ребенок.
Окулофарингеальная мышечная дистрофия
Окулофарингеальная мышечная дистрофия (OPMD) — это редкое генетическое заболевание, которое вызывает слабость мышц вокруг верхних век и части глотки, называемой глоткой. Дополнительная информация включает:
- Редко, поражает менее одного человека из 100000
- В равной степени влияет на всех полов
- Поражает в первую очередь глаза и горло
- Плечи, бедра и бедра также могут быть поражены
- Обычно начинается в возрасте от 40 до 60 лет
- Может вызывать такие осложнения, как проблемы со зрением, трудности с речью и глотанием, а также проблемы с подвижностью
Что такое спинальная мышечная атрофия?
Хотя спинальная мышечная атрофия звучит как мышечная атрофия, на самом деле это генетическое заболевание, симптомы которого имеют некоторое сходство с мышечной дистрофией.
Типы мышечной атрофии
Атрофия мышц обычно подразделяется на три типа:
Физиологические (неиспользованные)
Физиологическая атрофия мышц возникает в результате продолжительных периодов времени, когда мышцы не используются совсем или используются недостаточно. Другими словами, как гласит пословица, «используй или потеряй».
Хотя для значительной атрофии мышц может потребоваться время, признаки атрофии на молекулярном уровне могут появиться уже после недели снижения активности.
Существует множество причин, по которым может возникнуть физиологическая атрофия мышц, но некоторые из наиболее распространенных включают:
- Прикованность к постели
- Работа, требующая много сидения или небольшой активности
- Проблемы со здоровьем или состояния, ограничивающие движение или снижающие активность
- Неспособность двигать конечностями из-за состояния здоровья, например инсульта
- Все, что приводит к нерегулярной работе мышц
Физиологическая атрофия мышц может быть вызвана даже космическими путешествиями из-за отсутствия гравитации.
Этот тип мышечной атрофии часто можно обратить вспять, изменив образ жизни и увеличив физическую нагрузку, если потеря мышц была вызвана только неиспользованием.
Физиологическая атрофия мышц также может возникать как вторичный тип атрофии по сравнению с другими типами мышечной атрофии, когда они ограничивают движение и активность.
Патологический
Патогенная атрофия мышц может возникнуть в результате:
- Плохое питание или голодание
- Старение (называемое саркопенией)
- Заболевания, такие как болезнь Кушинга (в результате чрезмерного употребления кортикостероидных препаратов)
Нейрогенные
Нейрогенная атрофия поражает нервы, которые соединяются с мышечной тканью.Это наиболее серьезная форма мышечной атрофии.
Нейрогенная атрофия возникает, когда эти нервы повреждены или нервы поражены болезнью. Из-за повреждения нерва или нервов этот тип атрофии обычно необратим.
Некоторые состояния и заболевания, которые могут повлиять на нервы, контролирующие мышцы, включают:
- Боковой амиотрофический склероз (БАС, или болезнь Лу Герига)
- Синдром Гийена-Барре
- Рассеянный склероз
- Повреждение отдельного нерва, как при синдроме запястного канала
- Полиомиелит (полиомиелит)
- Травма спинного мозга
- Травма нерва
- Диабет
- Токсины, поражающие нервы
- Употребление алкоголя
Симптомы
Симптомы мышечной дистрофии-
Слабость и атрофия мышц
-
Затруднения при ходьбе, лазании, прыжках и других физических нагрузках
-
Телята увеличенные
-
Проблемы с сердцем
-
Проблемы с дыханием
-
Изогнутый корешок
-
Мышечная боль
-
Жесткие или свободные соединения
-
Затрудненное глотание
-
Веревочная походка
-
Слабость мышц
-
Потеря мышечной ткани
-
Затруднения с балансом
-
Проблемы с мобильностью
-
Онемение или покалывание
-
Мышечные подергивания, судороги, боли и боли
Симптомы мышечной дистрофии
Симптомы мышечной дистрофии зависят от типа, но обычно прогрессируют, становятся все более изнурительными и включают мышечную слабость и атрофию.
Мышечная дистрофия Дюшенна
Симптомы мышечной дистрофии Дюшенна включают:
- Прогрессирующая мышечная слабость и атрофия, начинающаяся в верхних частях ног и таза, затем распространяющаяся на верхние части рук
- Перевязывающая походка
- Потеря некоторых рефлексов
- Затруднения при вставании из положения лежа или сидя
- Трудности бега, прыжков и подъема по лестнице
- Изменения осанки
- Телята увеличенные
- Неуклюжесть и частые падения
- Нарушение дыхания
- Слабость легких
- Кардиомиопатия
- Респираторные инфекции
- Затруднения при глотании
- Истончение костей и сколиоз (искривление позвоночника)
- Когнитивные и поведенческие нарушения
Мышечная дистрофия Беккера
Симптомы мышечной дистрофии Беккера включают:
- Мышечная слабость, прежде всего в плечах, плечах, бедрах и тазу
- Ходьба на носках
- Частые падения
- Сложность подъема с пола
- Телята увеличенные
- Мышечные судороги
- Сердечные осложнения
- Когнитивные и поведенческие нарушения
Врожденная мышечная дистрофия
Симптомы врожденной мышечной дистрофии включают:
- Слабые мышцы
- Изогнутый корешок
- Слишком жесткие или ослабленные соединения
- Несоблюдение возрастных критериев двигательной функции и мышечного контроля
- Легкая или тяжелая дегенерация скелетных мышц
- Невозможность сидеть, стоять или ходить без опоры
- Респираторные заболевания
- Проблемы с глотанием
- Деформации стопы
- Возможные интеллектуальные нарушения
- Проблемы со зрением
- Проблемы с речью
- Изъятия
- Структурные изменения головного мозга
Дистальная мышечная дистрофия
Симптомы дистальной мышечной дистрофии включают:
- Слабость и атрофия мышц кистей, предплечий, голеней и стоп
- Обычно прогрессирует медленно, редко приводит к полной нетрудоспособности
- Затруднения с точным движением руки и разгибанием пальцев
- Трудности при ходьбе и подъеме по лестнице
- Неспособность прыгать или стоять на каблуках
Мышечная дистрофия Эмери-Дрейфуса
Симптомы мышечной дистрофии Эмери-Дрейфуса включают:
- Медленно прогрессирующая атрофия мышц плеча и голени
- Симметричная слабость
- Контрактуры (фиксированное напряжение мышц) позвоночника, лодыжек, коленей, локтей и задней части шеи
- Локти заблокированы в согнутом положении
- Жесткий позвоночник
- Ухудшение плеча
- Ходьба на носках
- Легкая слабость лица
- Проблемы с сердцем, обычно к 30 годам, часто требующие кардиостимулятора или другого вспомогательного устройства
- Прогрессирующая легочная или сердечная недостаточность
Facioscapulohumeral Muscular Dystrophy
Симптомы фасциально-лопаточно-плечевой мышечной дистрофии включают:
- Прогрессирующая мышечная слабость лица, плеч и предплечий
- Часто сначала поражаются мышцы вокруг глаз и рта, затем плечи, грудь и плечи
- Асимметричная слабость
- Внешний вид скошенных плеч и крылатых лопаток
- Снижение рефлексов
- Изменения во внешности лица (кривая улыбка, надутый вид, приплюснутые черты лица или маска)
- Неспособность сморщить губы или свистеть
- Затруднения при глотании, жевании или речи
- Респираторные заболевания
- Потеря слуха
- Аномальная кривая качания позвоночника
- Боль в пораженной конечности
Конечностно-поясная мышечная дистрофия
Симптомы мышечной дистрофии конечностей включают:
- Прогрессирующая потеря мышц и симметричное ослабление произвольных мышц, особенно в плечах и вокруг бедер
- Слабость в ногах и шее
- Перевязывающая походка
- Трудности вставать со стульев, подниматься по лестнице или переносить тяжелые предметы
- Частые падения
- Невозможность запуска
- Контрактуры мышц спины, придающие вид жесткости позвоночника
- Нарушение проксимальных (ближайших к центру тела) рефлексов
- Кардиомиопатия
- Респираторные осложнения
- Тяжелая форма инвалидности — обычное явление в течение 20 лет после начала болезни
Миотоническая мышечная дистрофия
Симптомы миотонической мышечной дистрофии включают:
- Трудность или неспособность расслабить мышцы после внезапного сокращения
- Слабость мышц лица и передней части шеи
- Хаггарда, лицо «топорик» и тонкая лебединая шея
- Атрофия и слабость мышц предплечья
- Сердечные осложнения
- Затруднения при глотании
- Птоз («опущенные» веки)
- Катаракты
- Нарушение зрения
- Раннее лобное облысение
- Похудание
- Эректильная дисфункция
- Атрофия яичек
- Легкая умственная отсталость
- Повышенное потоотделение
- Сонливость / избыточная потребность спать
- Нерегулярные менструации / бесплодие
Младенцы и дети с врожденной миотонической мышечной дистрофией могут проявлять:
- Затруднения при глотании или сосании
- Нарушение дыхания
- Отсутствие рефлексов
- Деформации и контрактуры скелета (например, косолапость)
- Слабость мышц (особенно в области лица)
- Психическое нарушение
- Задержка моторного развития
Окулофарингеальная мышечная дистрофия
Симптомы окулофарингеальной мышечной дистрофии включают:
- Опущение век (иногда сильное)
- Слабость лицевых мышц
- Слабость мышц глотки в горле
- Атрофия языка
- Затруднения при глотании
- Изменения голоса
- Двоение в глазах и проблемы с верхним взором
- Пигментный ретинит (прогрессирующая дегенерация сетчатки, которая влияет на ночное и периферическое зрение)
- Нарушения сердечной деятельности
- Слабость и атрофия мышц в области шеи и плеч, а иногда и конечностей
- Затруднения при ходьбе, подъеме по лестнице, стоянии на коленях или сгибании
Симптомы мышечной атрофии
Атрофия мышц — это симптом.Это просто означает потерю (или «истощение») мышечной ткани.
Симптомы, которые часто сопровождают атрофию мышц (особенно симптомы нервно-мышечных расстройств), включают:
- Мышечная слабость
- Потеря мышечной массы
- Подергивание, судороги, ломота и боли в мышцах
- Сложности передвижения
- Онемение, покалывание или болезненные ощущения
- Проблемы с глотанием
- Проблемы с дыханием
- Веки отвисшие
- Двойное зрение
- Проблемы с балансом
- Затруднения при ходьбе
- Водопад
- Слабость лица
- Потеря мышечной координации
- Постепенная потеря памяти
- Прогрессирующая потеря движения
Лечение
Лечение мышечной дистрофии-
Физиотерапия
-
Респираторная терапия
-
Логопед
-
Трудотерапия
-
Хирургия
-
Медикаментозная терапия
-
Генная терапия
Лечение мышечной дистрофии
Мышечную дистрофию нельзя предотвратить или вылечить, но существуют методы лечения с целью облегчения симптомов, улучшения качества жизни и замедления прогрессирования заболевания.Лечение включает:
- Физическая терапия : включает в себя физическую активность и упражнения на растяжку для сохранения гибкости и силы мышц
- Респираторная терапия : Лечение для предотвращения или задержки проблем с дыханием и, при необходимости, оборудование, такое как вентилятор, для помощи при дыхании
- Логопед : Помогает людям со слабостью лицевых мышц узнать, как максимизировать их мышечную силу, и предлагает средства коммуникации для тех, кто в этом нуждается.
- Трудотерапия : Помогает заново изучить утраченные двигательные навыки, работать с ослабленными мышцами для выполнения заданий, использовать личные вещи, такие как расческа и посуда, а также вспомогательные устройства, такие как инвалидное кресло
- Хирургия : Тип операции зависит от состояния, но некоторые операции включают установку кардиостимулятора, удаление катаракты или операцию на позвоночнике
- Медикаментозная терапия : Лекарства, используемые для лечения мышечной дистрофии, включают глюкокортикоиды (тип кортикостероидного гормона, который уменьшает воспаление), противосудорожные препараты (для помощи в контроле судорог и мышечных спазмов), иммунодепрессанты (чтобы помочь отсрочить некоторое повреждение умирающих мышечных клеток) , сердечные препараты, такие как бета-блокаторы и ингибиторы ангиотензинпревращающего фермента (АПФ)
- Генная терапия : изучаются методы восстановления способности гена вырабатывать полезные белки как способ лечения мышечной дистрофии
Лечение атрофии мышц
Лечение атрофии мышц часто включает лечение основного заболевания.Успех этих методов лечения также зависит от того, что вызывает атрофию.
Физиологическая атрофия часто хорошо поддается лечению и даже может быть купирована. Эти методы лечения включают:
- Упражнения, такие как тренировки с отягощениями
- Упражнения с малой ударной нагрузкой, такие как плавание и занятия водными видами спорта
- Повышенное потребление белка
- Привычки здорового образа жизни, такие как диета, сон и внимательность
- Реабилитационные упражнения, такие как физиотерапия, трудотерапия и пассивные движения (движения, выполняемые терапевтом)
- Упражнения со вспомогательными приспособлениями, такими как скобы или шины
Цель состоит в том, чтобы заставить эти мышцы двигаться и использовать их любым возможным способом.
Слово Verywell
Хотя мышечная дистрофия может вызвать атрофию мышц, это не одно и то же состояние.
Мышечная дистрофия — это генетическое заболевание, охватывающее девять основных типов, тогда как мышечная атрофия относится к потере мышечной ткани.